Next: Implementation Up: Metal CONQUEST Previous: ScaLAPACK Performance Profiling Contents
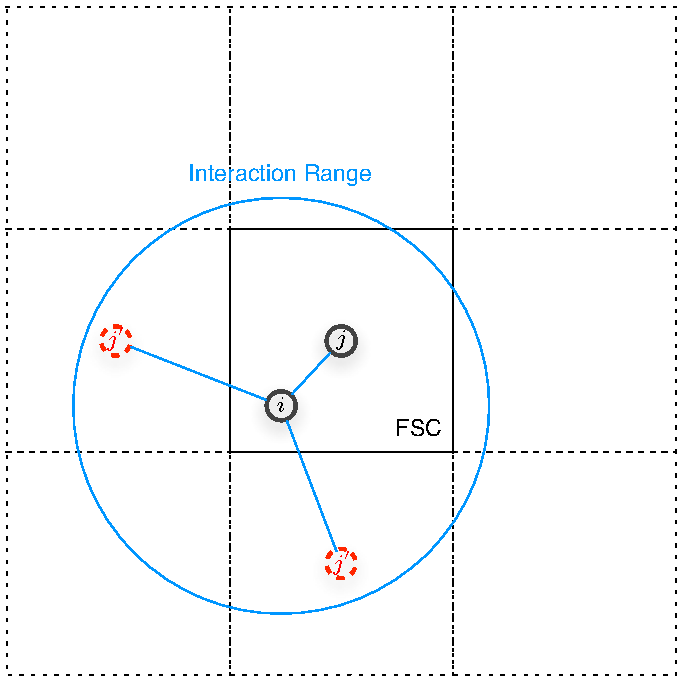 |
The CONQUEST implementation of matrices do not regard any system (bulk or otherwise) to have periodic boundary conditions, instead the code treats any location in real space as it is[5]. The cell from user input is regarded as the Fundamental Simulation Cell (FSC), and the FSC is repeated in all lattice directions so that all atoms taking part of interaction with that inside the FSC are taken account of (see figure 9). All quantum-mechanical operators are represented by matrices using support functions (which for the purpose of this report may be regarded as a set of basis functions upon which we have our matrix representation). For details on the meaning of support functions, how these are formed from the (actual) basis--which can either be Pseudo-Atomic Orbitals (PAOs) or B-Spline functions--and how are the quantum mechanical quantities represented by these support functions, please refer to [4,5].
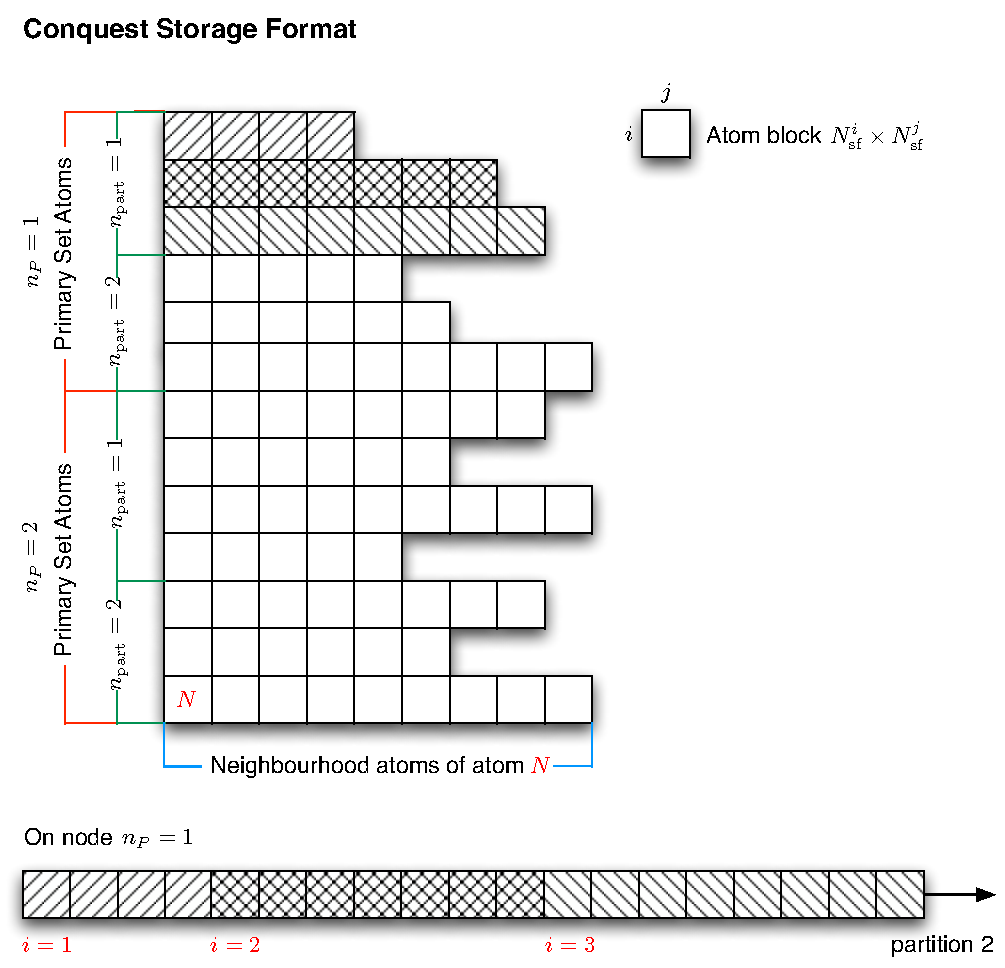 |
The CONQUEST method of storing matrices of the form
To update the electron density CONQUEST solves the
generalised eigen-value problem
It can be shown[3] that the hermitian matrices
![]() such as
such as
![]() and
and
![]() defined in equation
(16) and (17) can be calculated from the native
CONQUEST matrices using the relationship
defined in equation
(16) and (17) can be calculated from the native
CONQUEST matrices using the relationship
A call to ScaLAPACK subroutine pzhegvx is made to obtain the
set of eigenvalues (the band structure)
![]() which
are used to calculate the Fermi-energy and occupation function (this
is discussed briefly in section 3). Once this
is done another call to
which
are used to calculate the Fermi-energy and occupation function (this
is discussed briefly in section 3). Once this
is done another call to pzhegvx is made to get the eigenvectors
for each ![]() point, the new electronic density can than be
calculated using formula[3]
point, the new electronic density can than be
calculated using formula[3]
pzhegvx because we cannot calculate the density without knowing
the occupation function first, but on the other hand since the band
structure needs eigenvalues calculated for all pzhegvx all eigenvectors are
then need to be stored. By calling pzhegvx twice we can save
significant memory by simply accumulating the eigenvectors into the
density matrix. It was found2 also that
the call to pzhegvx for only calculating eigenvalues is only
about 10% the cost of the full call that also calculates the
eigenvectors.
The original CONQUEST implementation solves equation
(15) one ![]() point at a time. And the matrices
point at a time. And the matrices
![]() and
and
![]() are then mapped onto new
matrices
are then mapped onto new
matrices
![]() and
and
![]() distributed across all
available processors arranged in a BLACS processor grid according to
ScaLAPACK cyclic block format. The calculated eigenvectors from
ScaLAPACK for each
distributed across all
available processors arranged in a BLACS processor grid according to
ScaLAPACK cyclic block format. The calculated eigenvectors from
ScaLAPACK for each ![]() are then transfered from ScaLAPACK data
format and accumulated into
are then transfered from ScaLAPACK data
format and accumulated into
![]() stored across the
processors in CONQUEST format, and the self-consistent
calculation carries on from there.
stored across the
processors in CONQUEST format, and the self-consistent
calculation carries on from there.
We note that calculations involved for solving eigenvectors for
different ![]() are independent from each other. If we could add a
degree of freedom of allowing subgroups of processors working on
different
are independent from each other. If we could add a
degree of freedom of allowing subgroups of processors working on
different ![]() then it would allow us to choose better optimised
parameters for the ScaLAPACK calculations. For matrices of a given
size there is an optimal number of processors that should be allowed
to work on it, and too many processors means inefficient
communications taking over. Hence parallelising calculation in
then it would allow us to choose better optimised
parameters for the ScaLAPACK calculations. For matrices of a given
size there is an optimal number of processors that should be allowed
to work on it, and too many processors means inefficient
communications taking over. Hence parallelising calculation in
![]() would in theory allow one to use more processors more
efficiently by having groups of optimal number of processors working
for each ScaLAPACK subroutine call. Since for metallic calculations,
te number of
would in theory allow one to use more processors more
efficiently by having groups of optimal number of processors working
for each ScaLAPACK subroutine call. Since for metallic calculations,
te number of ![]() points required are in the order of 1000s, this
is a real degree of freedom we can exploit, especially for
calculations running on HPC systems such as HECToR.
points required are in the order of 1000s, this
is a real degree of freedom we can exploit, especially for
calculations running on HPC systems such as HECToR.