The most commonly used algorithms to self-consistently find the electron density
of a material, including the standard algorithms in the CRYSTAL code, scale as
the third power of the system size, i.e. 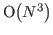 . In CRYSTAL, for large
systems, the bottle neck is the explicit diagonalisation of the Hamiltonian
(Fock or Kohn-Sham) matrix, an operation. For codes that do
not diagonalise the Hamiltonian, the cubic scaling occurs because
. In CRYSTAL, for large
systems, the bottle neck is the explicit diagonalisation of the Hamiltonian
(Fock or Kohn-Sham) matrix, an operation. For codes that do
not diagonalise the Hamiltonian, the cubic scaling occurs because 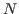 states
need to be found which are
states
need to be found which are 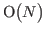 size and also must be orthogonal to the
other states. This unfavourable scaling limits traditional DFT computations
to systems with a maximum size of a few thousand of atoms. Computations for
systems larger than a few hundred atoms are uncommon.
size and also must be orthogonal to the
other states. This unfavourable scaling limits traditional DFT computations
to systems with a maximum size of a few thousand of atoms. Computations for
systems larger than a few hundred atoms are uncommon.
To enable the study of large systems, the scaling must be reduced.
Fortunately, the interaction between electrons in a many electron system is
almost always short ranged as the density matrix decays exponentially with
distance [7]. There have been several strategies developed
to exploit this near-sightedness'' of electrons in a variety of codes including
ONETEP [8], CONQUEST [9] and SIESTA
[10]. The divide and conquer algorithm chosen for this project
is described later in Section 2.
Daniel R. Jones 2011-12-06