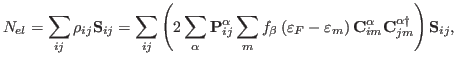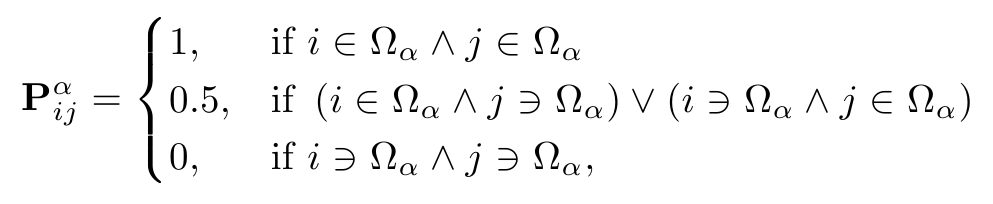Next: Proof of Principle Example Up: Implementation of a Divide Previous: Original dCSE Application Milestones Contents
|
More formally, a computation to calculate the single point energy of a system
![]() , whose single particle wavefunctions are written as a linear
combination of a localised basis set,
, whose single particle wavefunctions are written as a linear
combination of a localised basis set,
![]() , can be
approximately solved by dividing
, can be
approximately solved by dividing ![]() into a set of subsystems,
into a set of subsystems,
![]() , and defining a partition matrix
, and defining a partition matrix
| (3) |
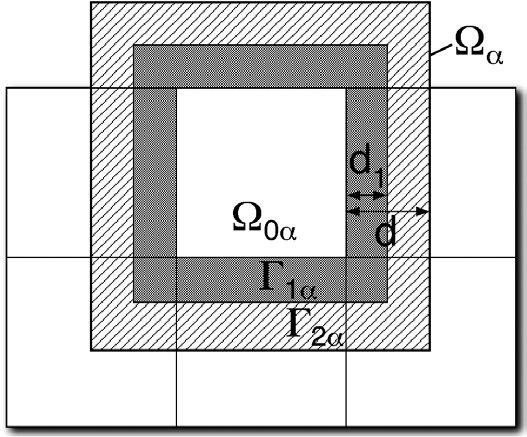
The accuracy of the electronic structure computed using this method varies depending on the partitioning of the system into subsystems. During this dCSE project, two methods for partitioning the system have been implemented. The simplest, most conservative method is for each subsystem to contain a single core atom. This method has been implemented in SIESTA [10] and is equivalent to a Mulliken population analysis. This method has a single parameter, the radius of the halo around the core atom. The appropriate size for the halo depends on the material being studied and can be defined in the CRYSTAL input file. The partition matrix for the system is defined as follows:
The alternative solution implemented in CRYSTAL (during this dCSE project) is
to leave the definition to the user and explicitly define each subsystem by
selecting a set of atoms (or ghost atoms) and defining the partition matrix in
atom-by-atom terms by hand. The way it has been implemented,
![]() must be identical for all basis functions localised on the same atom.
This allows users to have the flexibility to use the D&C algorithm in the
way that is most suited to their problem. The code which partitions the
systems into subsystems has been designed so that new heuristics for defining
the partition matrix can be added at a later date without altering the
structure of the code. The newly implemented D&C algorithm simply needs a
mapping array, which associates atoms in the subsystems with atoms in the
reconstructed system, and a partition matrix for each subsystem. The
must be identical for all basis functions localised on the same atom.
This allows users to have the flexibility to use the D&C algorithm in the
way that is most suited to their problem. The code which partitions the
systems into subsystems has been designed so that new heuristics for defining
the partition matrix can be added at a later date without altering the
structure of the code. The newly implemented D&C algorithm simply needs a
mapping array, which associates atoms in the subsystems with atoms in the
reconstructed system, and a partition matrix for each subsystem. The
![]() matrices defined must satisfy Equation
(1).
matrices defined must satisfy Equation
(1).
In the time available, point 4 on the list of milestones has not yet been addressed. The long range electrostatics of the system have been discarded, for systems that are largely non-polar, this should make no difference and the electrostatics included by the interaction of the core with the halo region should be sufficient, as will be shown in the next section.
Daniel R. Jones 2011-12-06