Optimisation of ONETEP code
Dr Nicholas D.M. Hine and Dr Peter D. Haynes, Imperial College
Dr Arash A. Mostofi (Imperial), Dr Chris-Kriton Skylaris (Southampton)
and Prof Mike C. Payne (Cambridge)
Figure 1. 4375-atom Nanorod of Wurtzite-structure Gallium Arsenide.
ONETEP (Order-N Electronic Total Energy Package) is a linear-scaling code for quantum-mechanical simulations based on density functional theory, developed jointly at Cambridge, Imperial College and Southampton University. ONETEP can perform ab initio electronic structure calculations where the total computational effort of the calculation scales only linearly with system size, as opposed to the cubic scaling of traditional methods. It can therefore be used to simulate systems of thousands or tens of thousands of atoms, as opposed to the hundreds to which traditional DFT methods are limited. ONETEP combines this favourable scaling with a systematically improvable basis set, allowing for highly-accurate calculations.
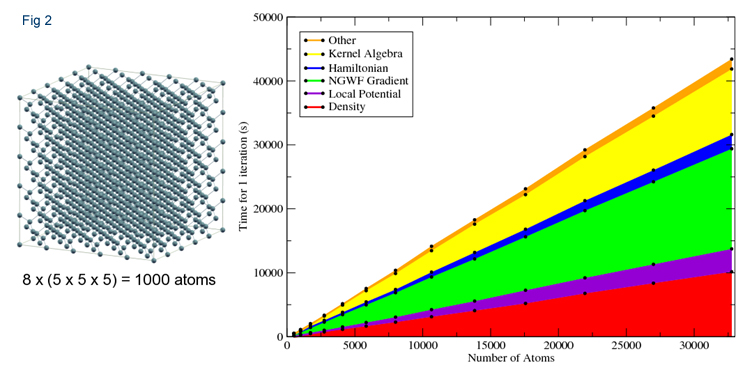
Recent development work using HECToR and Imperial's CX1 machine has greatly improved the parallel efficiency of ONETEP, especially when studying systems of solids. Recently, calculations involving systems of up to 32,768 of crystalline silicon have been demonstrated (Figure 2) and scaling efficiently up to over 256 cores.
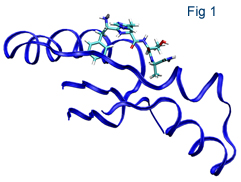
ONETEP is currently being used for a wide variety of applications by several UK and international research groups, including studies of protein-ligand interactions (Figure 3) and self-assembly in semiconductor nanorods (Figure 1). ONETEP is available for research under an Academic License and is marketed by Accelrys as part of the Materials Studio package.
The work was principally carried out at Imperial College, by Dr Nicholas D.M. Hine and Dr Peter D. Haynes, in collaboration with the rest of the ONETEP Developers Group: Dr Arash A. Mostofi (Imperial), Dr Chris-Kriton Skylaris (Southampton) and Prof Mike C. Payne (Cambridge).
The results of the optimisation work can be found in the following paper "Linear-scaling density-functional theory with tens of thousands of atoms: Expanding the scope and scale of calculations with ONETEP, N.D.M. Hine, P.D. Haynes, A.A. Mostofi, C.-K. Skylaris and M.C. Payne, Computer Physics Communications 180, 1041, (2009)." doi:10.1016/j.cpc.2008.12.023
The ONETEP project has a website at http://www.onetep.org.
Figure 2. Scaling of computational time with number of atoms for crystalline Silicon.
Figure 3. Candidate ligand structure interacting with fragment of protein.