Materials Chemistry Consortium Highlights
HPC Materials Chemistry Consortium (MCC) embraces various institutions in the UK - : University College London, Imperial College London, University of Bath, University of Birmingham, University of Cambridge, Cardiff University, King’s College London, The University of Sheffield, Trinity College Dublin, University of York. MCC is chaired by Prof Richard Catlow.
MCC has played a major role in UK computational science and, since its foundation in 1994, has exploited the latest developments in HPC technologies, in a wide ranging programme of development, optimisation and applications studies aimed at modelling and predicting the structures, properties and reactivities of functional materials, including catalysts, ceramics, minerals and molecular materials. The programme embraces both large scale simulations based on forcefields and electronic structure techniques employing Density Functional Theory, Hartree Fock and hybrid techniques. Strong emphasis is placed on code development and optimisation for MPP platforms while several applications highlight systems of industrial importance. There is strong symbiosis between the modelling studies of the consortium and experimental programmes.
Inside the consortium, there are seven main themes grouping all the projects that request computing time on HECToR; each theme has a leader indicated in parenthesis:
- Biomaterials (Prof Nora de Leeuw)
- Environment (Prof John Harding)
- Materials for Energy Technology (Prof Saiful Islam)
- Nano and Defect Chemistry (Prof Alex Shluger)
- Quantum Devices (Prof Nicholas M. Harrison)
- Reactivity (Dr David Willock )
- Surfaces and Interface (Dr Ben Slater)
Insights could lead to better rechargeable batteries
A. R. Armstrong, C. Lyness, P. M. Panchmatia, M. S. Islam and P. G. Bruce
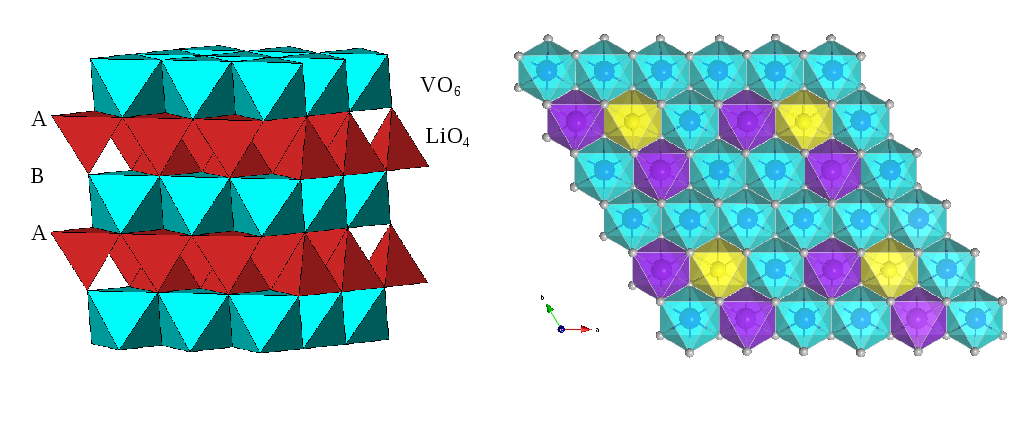
Recent research on new materials which could lead to lithium batteries storing more energy has been published in the leading journal Nature Materials.
Rechargeable lithium batteries have helped power the 'portable revolution' in mobile phones, laptops and mp3 players.
New generations of lithium batteries are now being developed for hybrid electric vehicles and for energy storage of wind/solar power to help cut carbon emissions.
Now the research groups of Prof Saiful Islam of the Department of Chemistry at the University of Bath, and Prof Peter Bruce at the University of St. Andrews, have gained important insights into novel oxide compounds to help lithium batteries become more efficient.
Recent reports that lithium can insert or "intercalate" into a novel lithium vanadium oxide at an unprecedented low voltage, represent an important milestone in this field.
These oxides can store more electrical energy than the current graphite electrode.
The research team used a powerful combination of structural experiments and DFT computer simulations on Hector to unravel, for the first time, why excess lithium in these compounds “switch on” lithium intercalation.
Prof Saiful Islam said "Such understanding is important for the future design and development of next generation batteries for electric vehicles."
Related links:
- Prof Saiful Islam webpage
- The lithium intercalation process in the low-voltage lithium battery anode Li1+xV1−xO2.
- A. R. Armstrong, C. Lyness, P. M. Panchmatia, M. S. Islam and P. G. Bruce, Nature Materials 10, 223–229 (2011)
Fluorine Environment in Bioactive Glasses: ab initio Molecular Dynamics Simulations
J. K. Christie, A. Pedone, M. C. Menziani, and A. Tilocca
Fluorinated bioactive glasses combine the antibacterial properties of fluorine, with the bone-bonding capability of bioactive glasses. They could be used in dentistry by bonding to the tooth or bone, and slowly releasing fluorine into the mouth, helping to protect the teeth. The release of fluorine from the glass depends on its local structure, and very accurate first-principles simulations were used to identify this, and resolve some of the uncertainties from experimental work on this glass. Fluorine bonds almost entirely to sodium and calcium ions in the glass, which causes the formation of regions in the glass which are rich in these ions and lack certain others (silicon and phosphorous), which affects the biological activity of the glass.
Related link:
- Fluorine Environment in Bioactive Glasses: ab Initio Molecular Dynamics Simulations J. K. Christie, A. Pedone, M. C. Menziani, and A. Tilocca, J. Phys. Chem. B 115, 2038 (2011)
Thermodynamic Stability of LaMnO3 and its Competing oxides: A Hybrid Density Functional Study of an Alkaline Fuel Cell Catalyst
E. A. Ahmad, L. Liborio, D. Kramer, G. Mallia, A. R. Kucernak and N. M. Harrison
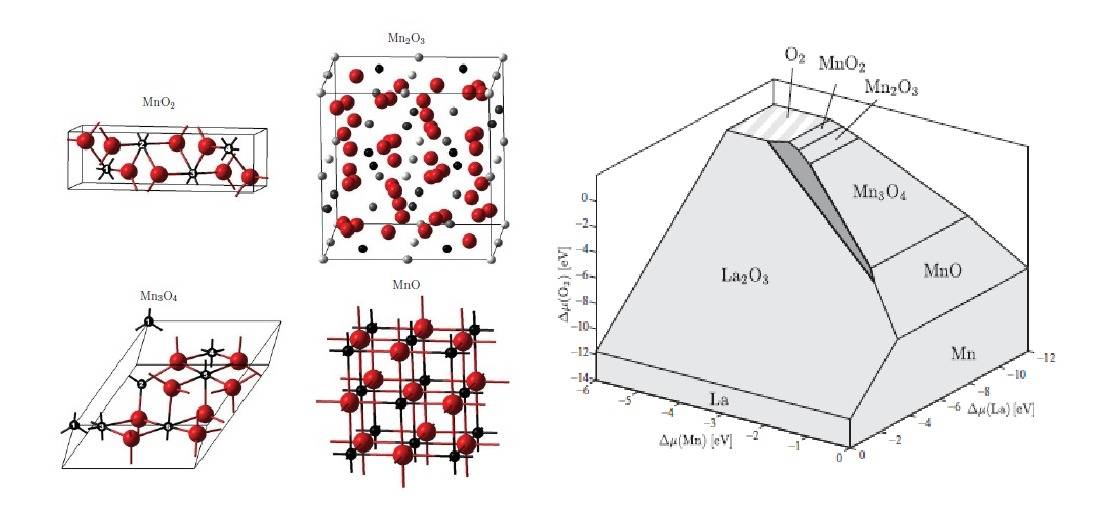
LaMnO3 is well known as a perovskite material that exhibits useful properties for magnetic sensors and solid oxide fuel cells, but recent work has shown that it is also a promising candidate for facilitating oxygen reduction in alkaline fuel cells (AFCs). This presents a valuable opportunity for commercialization of AFC technology, since LaMnO3 has a significant economic advantage over conventional fuel cell catalysts such as platinum.
In order to understand this complex material a group of researchers from the Thomas Young Centre, London Centre of technology, Imperial College Chemistry Department, Daresbury Laboratory and Southampton University have carried out a theoretical study of the thermodynamics of bulk LaMnO3 and its related compounds. They have succeeded in establishing a framework for accurate study of the catalytic LaMnO3 surface and have demonstrated a practical methodology within which quantum mechanics can be used to compute the phase stability of a complicated transition metal oxide reliably.
Related links:
- Thermodynamic Stability of LaMnO3 and its competing oxides: A Hybrid Density Functional Study of an Alkaline Fuel Cell Catalyst.
- E. A. Ahmad, L. Liborio, D. Kramer, G. Mallia, A. R. Kucernak and N. M. Harrison, Phys. Rev. B, 84, 085137 (2011)
Oxide-metal interactions from ab initio calculations
R. Grau-Crespo and N. H. de Leeuw

Oxide-supported gold catalysts are known to be very active in the low-temperature water gas shift reaction and in the preferential oxidation of CO in the presence of hydrogen, which are important reactions for the purification of hydrogen feeds in fuel cells.
We have used electronic structure calculations based on the density functional theory to investigate the interaction of gold atoms with the surfaces of cerium and zirconium oxides. We have shown that gold adatoms at the (111) surface of ceria can adopt Au0, Au+ or Au- electronic configurations (see figure above) depending on the adsorption site, thus modifying the redox properties of the ceria surface [1]. The modelling of this interaction is complicated by the self-interaction and volume overestimation errors in traditional DFT calculations, which critically affect the relative stabilities of the different electronic configurations of the adatoms [2].
On the other hand, the adsorption of gold at non-defective zirconia surfaces does not lead to any charge transfer behavior [3], but we have found that the substitution of gold in lattice positions at a zirconia surface induces a dramatic change in the redox properties of the surface, which becomes easily reducible thanks to Au(III) - Au(I) transitions [4]. Pd adatoms can also transfer charge to the Ce atoms at the ceria surface, according to our calculations. Similar conclusions have been reached by an XPS investigation of Pd deposition on ceria ultra-thin films [5]. This work has led to a better understanding of oxide-metal interactions, in particular for systems of interest in catalysis.
Related links:
- Electronic charge transfer between ceria surfaces and gold adatoms: a GGA+U investigation. N. C. Hernandez, R. Grau-Crespo, N. H. de Leeuw and J. F. Sanz., Physical Chemistry Chemical Physics 11, 5246 - 5252 (2009).
- Theoretical investigation of the deposition of Cu, Ag and Au atoms on the ZrO2(111) surface. R. Grau-Crespo, N. Cruz-Hernández, J. F. Sanz and N. H. de Leeuw. Journal of Physical Chemistry C 111, 10448 - 10454 (2007)
- On the difficulties of present theoretical models to predict the oxidation state of atomic Au adsorbed on regular sites of CeO2(111). M. M. Branda, N. J. Castellani, R. Grau-Crespo, N. H. de Leeuw, N. C. Hernandez, J. F. Sanz, K. M. Neyman and F. Illas. Journal of Chemical Physics 131, 094702 (2009).
- Redox Properties of Gold-Substituted Zirconia Surfaces. R. Grau-Crespo, N. Cruz-Hernández, J. F. Sanz and Nora H. de Leeuw. Journal of Materials Chemistry 19, 710-717 (2009).
- Redox behavior of the model catalyst Pd/CeOx/Pt(111). E. L. Wilson, R. Grau-Crespo, C. L. Pang, G. Cabailh, Q. Chen, J.A. Purton, C.R.A. Catlow, W. A. Brown, N. H. de Leeuw and G. Thornton. Journal of Physical Chemistry C, 112, 10918-10922, (2008).
An Apatite for Ion Conduction
P. M. Panchmatia, A. Orera, G. J. Rees, M. E. Smith, J. V. Hanna, P. R. Slater, and M. S. Islam
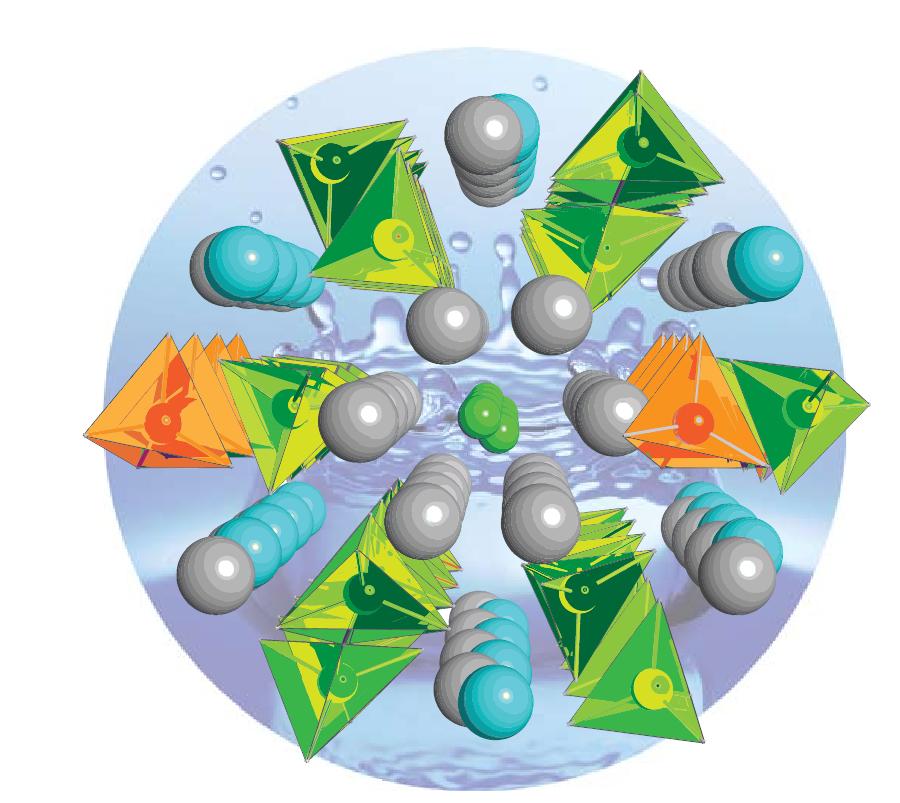
The viability of low carbon energy technologies such as fuel cells is crucially dependent on the fundamental advances in the component materials.
Si- and Ge-apatite compounds are attracting considerable interest as new fast oxide-ion conductors for use in solid oxide fuel cells (SOFCs).
Recently, Saiful Islam (Bath) with Peter Slater (Birmingham) and Mark Smith (Warwick) have utilised a powerful combined nuclear magnetic resonance (NMR) and computational approach to elucidate, for the first time, the oxygen defect sites and conductivity mechanisms in the novel apatite compound La8Y2Ge6O27, which exhibits high oxide-ion conductivity.
Through large-scale MD and DFT simulations on Hector and solid state 17O NMR studies they show that interstitial oxygen leads to five-coordinate Ge, and that oxide-ion migration occurs via cooperative mechanisms with evidence of a novel 'SN2-like' conduction mechanism.
The results are therefore of great significance in the search for new oxide ion conductors for clean energy applications.
Related links:
- Prof Saiful Islam webpage
- Oxygen Defects and Novel Transport Mechanisms in Apatite Ionic Conductors: Combined 17O NMR and Modeling Studies P. M. Panchmatia, A. Orera, G. J. Rees, M. E. Smith, J. V. Hanna, P. R. Slater, and M. S. Islam, Angew. Chemie. 50, 9328-9333 (2011)
He-atom scattering from MgO(100): A periodic quantum mechanical study.
R. Martinez-Casado, G. Mallia and N. M. Harrison. In collaboration with the Theoretical Chemistry Group of the University of Turin (Dr S. Casassa and Dr L. Maschio) and with the Institute of Physical and Theoretical Chemistry of the University of Regensburg (Dr D. Usvyat and Prof M. Schuetz).
He-atom scattering is potentially an important tool for the analysis of surface structure and dynamics. The technique is not complicated by surface charging and damage, which affect electron diffraction, microscopy and spectroscopy methods, and is therefore particularly useful in the study of insulating surfaces, such as MgO, TiO2 and ZnO. The use of He-atom scattering has, however, an important limitation, namely the difficulties involved in the interpretation of the experimental diffraction patterns due to the lack of a detailed understanding of the scattering potential and process. A first principles He-surface interaction potential is difficult to obtain as the widely used approximations based on density functional (DFT) and Hartree-Fock (HF) theory provide a poor treatment of weak inter-molecular dispersion interactions. Empirical corrections to DFT which approximate dispersion can be used but prove to be unreliable in many interesting systems and the alternative of using high quality quantum chemical methods based on clusters can often be complicated by issues related to convergence with respect to cluster size and the expense of the calculations. As an alternative an ab initio interaction potential has been computed using periodic local Moller-Plesset perturbation theory at second order (L-MP2), as recently implemented in the CRYSCOR program. HF+MP2 is shown to produce a qualitatively correct description of the long range dispersion interaction for He-MgO(100) but to underestimate the magnitude of the interaction. A quantitatively correct energy surface which reproduces the observed He binding energy, bound state energy levels and diffraction pattern can be obtained by scaling the long range attractive potential by 1.65 - a factor correcting for the MP2 underestimate of the correlation energy that is well known from quantum chemical calculations on noble gas dimers. A similar energy surface can be obtained by improving the underlying HF description of the electronic structure using hybrid exchange density functional theory before applying MP2 theory - the so-called HF+MP2(B3LYP) approximation. This work has demostrated for the first time that the problem of the interpretation of He-atom scattering experiments can be overcome through the application of efficient and reliable first principles theory.
Related links:
- Periodic quantum mechanical simulation of the He-MgO(100) interaction potential R. Martinez-Casado, G. Mallia, D. Usvyat, L. Maschio, S. Casassa, M. Schuetz and N. M. Harrison, J. Chem. Phys., 134, 014706 (2011)
- An alternative approach for the calculation of correlation energy in periodic systems: a hybrid MP2(B3LYP) study of the He-MgO(100) interaction R. Martinez-Casado, G. Mallia and N. M. Harrison, Chem. Commun., 47, 4385 (2011)
- He-atom scattering from MgO(100): Calculating Diffraction Peak Intensities From First Principles. R. Martinez-Casado, G. Mallia, D. Usvyat, L. Maschio, S. Casassa, M. Schuetz and N. M. Harrison, Phys. Chem. Chem. Phys., 13, 14750 (2011)
- Ab initio calculation of the MgO(100) interaction with He and Ne: a HF + MP2 and HF + MP2(B3LYP) comparison R. Martinez-Casado, G. Mallia and N. M. Harrison, Chem. Commun., 47, 11630 (2011)
Looking for the Origin of Life: Bio-mimesis for CO2 activation
Alberto Roldan-Martinez and N. H. de Leeuw

Primordial metabolism outgrowth may be postulated as a continuum from Earth’s geochemical processes to chemoautotrophic biochemical procedures on a sulphide mineral surface. Moreover, metal sulphides play a crucial role in the enzymes of some extant life forms. Thus, by mimicking enzyme activity, mineral surfaces may catalyse simple reactions yielding organic molecules from inorganic ones. At the onset of Life, the production of these pre-biotic molecules were possibly driven by super-hot hydrothermal vents deep in the ancient oceans or by ultraviolet light coming from beyond the precarious atmosphere. One of these atmospheric components was CO2, the same molecule which is a major contributor to the Climate Change on our planet. This study is concerned both with gaining insight into one of the most intriguing questions – the Origin of Life - and with finding a viable route to use this greenhouse gas as a raw material and yield useful organic feedstock molecules. Accurately modelling potential catalysts may provide decisive clues, not only in terms of reaction conditions or chemoautotrophic mechanisms, but also on how to improve the catalyst to enhance rates of production of a particular product. The use of the density functional theory (DFT) approach for surface reactions has already been employed extensively. Although this methodology is computationally expensive when applied to large and complex systems, the HPC facilities provided by HECToR now make this a feasible scenario.