The molecular dynamics algorithm solves Newton's equation of motion for a set of discrete molecules.
The interaction potential in the simplest case is the Lennard-Jones potential, but the solver can also model long-chain molecules.
Algorithmic acceleration is achieved via updated cell lists, which maintain a record of molecules within physical proximity.
These cells are optimised in order to maximise the number of steps where interacting molecules are within the same or neighbouring cells, before the need to reconstruct the cell list.
Further acceleration is achieved via neighbour lists which maintain a record of neighbouring molecules.
Parallelism is implemented via MPI domain decomposition.
The physical domain is divided among MPI processes, in a similar manner to the continuum solver.
Neighbouring cells, and hence neighbouring molecules, are therefore hosted on the same MPI process.
Halos, or duplicate copies of cells, at the boundaries are employed in order to maximise the number of computations prior to rebuilding of the cell and neighbour lists.
It should be noted that the domain decomposition parallelism in
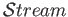
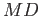 is similar to the approach adopted for
is similar to the approach adopted for
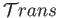
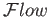 .
This facilitates the task of interfacing the two applications since a logical connectivity can be readily established between boundary processes at the continuum-MD boundary.
.
This facilitates the task of interfacing the two applications since a logical connectivity can be readily established between boundary processes at the continuum-MD boundary.
The performance of
was compared to LAMMPS which was developed by Sandia National Laboratories and was optimised and extensively tested for parallel performance.
The parallel scaling efficiency of
was shown to be similar to LAMMPS.
In addition, similar to
, the scalability of
has been verified extensively with target operation at 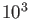 CPU cores.
Using
CPU cores.
Using
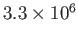 molecules,
shows 94% parallel efficiency on 1024 cores relative to the 8 cores performance on HECToR.
molecules,
shows 94% parallel efficiency on 1024 cores relative to the 8 cores performance on HECToR.
2013-05-31