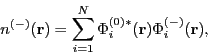Milestone 2 - This will comprise much of the framework of a releasable implementation of TDDFT using Hutter's published method. Issues of data distribution and parallel efficiency will not be addressed at this stage and an `off-the-shelf' eigensolver will be used. Initial testing and debugging. Code will be benchmarked against a set of existing calculations on molecular systems chosen from scientific literature. This work comprises stages 3-5 of the work plan.
Hutter reformulated the equations of time-dependent Hartree-Fock (TDHF) theory (applied to TDDFT with pure density functionals) such that they could be efficiently implemented in a plane-wave basis set. The TDHF equations are a non-Hermitian eigenvalue equation, which gives the excitation energies directly
| (3) |
In the context of a plane-wave code, the vectors ![]() and
and ![]() are the plane-wave coefficients of electronic wavefunctions, i.e.
are the plane-wave coefficients of electronic wavefunctions, i.e.
![]() from above. In CASTEP, these are four-dimensional arrays, the dimensions being G-vectors (plane-waves), k-points, bands and spin. For a TDDFT calculation, one typically has
from above. In CASTEP, these are four-dimensional arrays, the dimensions being G-vectors (plane-waves), k-points, bands and spin. For a TDDFT calculation, one typically has
![]() G-vectors,
G-vectors, ![]() bands, a single k-point and spin of 1 or 2 dpending on whether the system is being treated as spin-degenerate. Of course, storing and diagonalising a matrix of this size is innapropriate, especially as one is only interested in the lowest few excited states. Instead we turn to iterative methods, where only the result of the operator on a vector is required.
bands, a single k-point and spin of 1 or 2 dpending on whether the system is being treated as spin-degenerate. Of course, storing and diagonalising a matrix of this size is innapropriate, especially as one is only interested in the lowest few excited states. Instead we turn to iterative methods, where only the result of the operator on a vector is required.
Hutter defines this operator in equation 35 of his paper, where he chooses to separate it into two components, such that
![]() . The first contribution is from Kohn-Sham orbital energy differences
. The first contribution is from Kohn-Sham orbital energy differences
| (4) |
| (5) |
| (7) |
Given the effect of the ![]() operator on a given wavefunction, an `off-the-shelf' iterative eigensolver with a reverse communication interface can be used. By using a library routine, we can thoroughly test our implementation of the operator. We used two different solvers, namely ARPACK3 and EA19, the latter being from the HSL (formerly the Harwell Subroutine Library)4. ARPACK is written in Fortran77 and implements the Arnoldi process. HSL-EA19 is written in F95 + TR 15581 and implements a Jacobi-conjugate preconditioned gradients scheme.
operator on a given wavefunction, an `off-the-shelf' iterative eigensolver with a reverse communication interface can be used. By using a library routine, we can thoroughly test our implementation of the operator. We used two different solvers, namely ARPACK3 and EA19, the latter being from the HSL (formerly the Harwell Subroutine Library)4. ARPACK is written in Fortran77 and implements the Arnoldi process. HSL-EA19 is written in F95 + TR 15581 and implements a Jacobi-conjugate preconditioned gradients scheme.