Molecular Dynamics Simulation Of Multi-Scale Flows on GPUs
A dCSE project
S. Mulla, Dr S. Ivekovic and Prof. J. Reese
James Weir Fluids Lab,
University of Strathclyde,
Glasgow, UK
23 July 2012
Abstract
The aim of the project described in this report was to integrate the Molecular Fluid Dynamics simulation package OpenFOAM with the molecular-modelling library OpenMM, in order to enable the execution of Molecular Fluid Dynamics simulations on Graphics Processing Units (GPUs). The reason for this integration, rather than a straightforward substitution of OpenFOAM with OpenMM, was to combine the pre- and post-processing functionality of OpenFOAM (running on CPUs) with fast, GPU-based, Molecular Dynamics implementation provided by OpenMM. The result was a GPU-accelerated Molecular Fluid Dynamics simulation package with a pre- and post-processing capability.
Running engineering time- and length-scale Molecular Dynamics simulations entails a considerable computational effort and currently requires computational times that are prohibitively long for transformational advances in fluids engineering to be possible. GPU-accelerated Molecular Fluid Dynamics will enable faster and longer simulations, as well as better exploitation of computational resources. Not only does running simulations on GPUs relieve existing, CPU-based, high-performance computational resources, but it also enables simulations to be run on personal workstations equipped with suitable GPUs, thereby saving on large CPU-cluster waiting times and execution costs.
The emphasis of this project was on nano-scale fluid dynamics. The resulting software package will thus be of particular relevance to researchers interested in flows at the smallest scales.
Contents
- Introduction
This report describes the outcomes of a project conducted to accelerate the Molecular Dynamics simulations needed to perform research in nano-scale flows of engineering interest.
The tool of choice for running such simulations at the start of this project was the OpenFOAM library5 (Section 2.1). OpenFOAM had already been parallelised using the Message Passing Interface (MPI) framework2 and the MPI implementation was regularly used to run simulations on the in-house high-performance computer, ARCHIE-WeSt3. However, despite the MPI capability, simulations on engineering time- and length-scales still took prohibitively long to complete. To illustrate this, Figure 1 shows a graph of the performance of the standard OpenFOAM Molecular Dynamics solver at the start of the project. The graph shows the duration in seconds of a Molecular Dynamics simulation time step over a fluid system as a function of the number of Intel Xeon X5650 2.66 GHz CPU cores (with 12 cores per node) used to perform the simulation with MPI on ARCHIE-WeSt. The simulation is of a simple 3D box with periodic boundary conditions and containing 382,228 argon atoms.
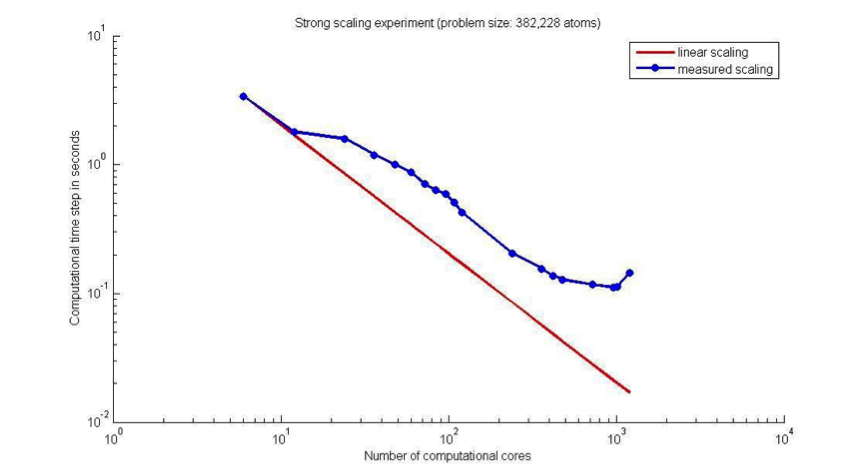
Figure 1: Strong scaling of the OpenFOAM MD solver on ARCHIE-WeSt (Intel Xeon X5650 2.66 GHz).
It can be seen from this figure that the computational time per MD time step decreases with the increasing number of CPU cores until the number of cores exceeds, in this particular case, 960. At this point, the MPI communication time exceeds that of the computation time per CPU core and it becomes counter-productive to split the computation between an even greater number of cores. The computational time per time step that was achieved with 960 cores was 0.1118 s. With the Molecular Dynamics time step of 5.4015 fs, the fastest we could simulate 5.4015 ns of problem time on a high-performance computer with OpenFOAM was on 960 cores, and this would take more than 31 hours.
Simulation of problem times of engineering interest is currently prohibitive, not only because we are not the sole users of the high performance computer and therefore cannot regularly request 960 cores, but also because, e.g., 5 microseconds of problem time for even this simple fluid problem would require 3.5 years to simulate at this computational speed. Longer time-scales (seconds, minutes) are well out of reach.
- Hybrid Solver (OpenFOAM, accelerated with OpenMM)
In order to reduce the computational time of running Molecular Dynamics simulations, this project aimed to accelerate the simulations by porting the simulation code to the GPU platform. An existing GPU-based molecular modelling library, OpenMM4 (Section 2.2), was chosen to help achieve this aim.
The strategy adopted during this project was to substitute the CPU-based Molecular Dynamics functionality of OpenFOAM with the equivalent OpenMM GPU-based functionality in such a way that the end user need not be aware of the modification in order to continue running MD simulations in OpenFOAM. This was done by keeping the OpenFOAM framework as the front end of the accelerated simulation interface, meaning that the pre- and post-processing capabilities, as well as the simulation setup tools of OpenFOAM, could continue to be used while the actual Molecular Dynamics algorithm was executed on a GPU platform, using the (appropriately extended) OpenMM library.
To achieve this, two main implementation stages were required. In the first stage, the core OpenFOAM Molecular Dynamics algorithm was replaced with the equivalent part of the OpenMM toolkit by using the OpenMM C++ API. In the second stage, the OpenMM functionality was extended in order to match that of the CPU-based OpenFOAM by adding fluid dynamics control and measurement tools. The final result was a GPU-accelerated simulation package for Molecular Fluid Dynamics with OpenFOAM as a front end - called the hybrid solver. An additional outcome of the project is a stand-alone OpenMM library with added control and measurement tools, which can be used either independently or to accelerate other simulation packages.
2.1 OpenFOAM
OpenFOAM5,7 is an open-source Computational Fluid Dynamics (CFD) C++ software package, developed by OpenCFD Ltd at the ESI group. In addition to its core CFD functionality, it also contains a Molecular Dynamics (MD) module which enables fluid dynamics simulations at the nano-scale. This module was initiated, and is being actively developed, by researchers in the James Weir Fluids Lab1 at the University of Strathclyde. In its standard MPI implementation, OpenFOAM enables CFD and MD simulations to be run on single and multiple CPUs. Strong scaling is effective for up to 1000 CPU cores, after which the communication bottleneck reduces the efficiency of the parallelised code. At the start of this project, MPI implementation of the OpenFOAM MD module was the main tool for running nano-scale fluid dynamics simulations in the James Weir Fluids Lab.
2.2 OpenMM
OpenMM4,6,8,9 is an open-source molecular simulation C++ toolkit developed by the Simbios National Center for Biomedical Computation and supported by the US National Institutes of Health. It has been developed with a strong emphasis on hardware acceleration, and greatly facilitates running general-purpose Molecular Dynamics (MD) simulations on GPUs. The toolkit uses C++, Cuda and OpenCL and currently allows accelerated MD simulations to be run on NVidia and AMD graphics cards, as well as on CPUs. The focus of the toolkit's functionality is on biomedical simulation; nevertheless, its well-designed, layered architecture allows for the necessary code extensions to be made in a structured way.
- Hybrid Solver Implementation Steps
As stated above, the goal set during this project was to embed the GPU functionality provided by the OpenMM library into the software architecture of OpenFOAM in such a way that the pre- and post-processing capability and the simulation-setup interface of OpenFOAM could continue to be used.
This goal was achieved by using the OpenFOAM interface to set up and initialise the simulation and then automatically extracting the necessary information to initialise and run an equivalent Molecular Dynamics simulation in OpenMM. The OpenMM library took care of running the simulation as well as performing the necessary measurements and control. When the simulation finished, the data structures and execution control were returned to OpenFOAM for post-processing.
In order to achieve the proposed OpenFOAM-OpenMM integration, a number of consecutive implementation steps were identified. These helped guide the project to its completion and are briefly summarised next.
3.1 Choice of accelerator platform
The OpenMM library GPU implementation is available in two sub-libraries (also called platforms): CUDA and OpenCL. When this project began, the OpenCL sub-library was more advanced and computationally much more efficient than its CUDA counterpart. At the same time, the OpenMM development team announced they would support OpenCL more actively in the future, and hence the OpenCL sub-library was adopted also for this project.
3.2 Delegation of the pairwise force calculations to GPU
The first step involved linking the OpenMM OpenCL platform with OpenFOAM through the OpenMM API and delegating the pairwise force calculations to OpenMM. All the remaining Molecular Dynamics algorithm steps continued to be executed in OpenFOAM. At every time step, molecular positions were communicated from OpenFOAM to OpenMM and pairwise forces were communicated back from OpenMM to OpenFOAM.
3.3 Port of the particle trajectory integration algorithm to OpenMM
Once the pairwise force calculation step was successfully delegated to OpenMM's OpenCL platform, the next step was to delegate also the particle trajectory integration. This enabled the core Molecular Dynamics algorithm to be completely executed on the OpenMM GPU platform.
3.4 Implementation of the Molecular Dynamics measurement tools in OpenMM
The Molecular Dynamics simulations that are run in the James Weir Fluids Lab use a variety of runtime measurement tools during the simulation run. Most of these measurements were not available in the OpenMM library. In this step, the runtime measurements were added to the OpenMM library.
3.5 Implementation of the Molecular Dynamics control tools in OpenMM
The OpenFOAM Molecular Dynamics module uses control tools, such as the Berendsen thermostat, which were not available in OpenMM. In this step, the missing control tools were implemented in OpenMM.
3.6 Implementation of fine-grained control and measurement tools
An important aspect of the measurement and control process is to be able to partition the simulation domain into bins and then to apply the control and measurement tools in each individual bin. In this step, the measurement and control tools that were added to the OpenMM library in Steps 3.4 and 3.5 were extended to work in bins.
- Work Packages
The starting point of the project described in this report was a Molecular Dynamics algorithm running in MPI-enabled OpenFOAM on one or multiple CPUs and an OpenMM 3.1 CUDA-based hybrid CPU-GPU solver with force calculations - but not the remainder of the MD algorithm - performed on the GPU. The end point was the OpenFOAM Molecular Dynamics algorithm ported to the GPU platform. This was achieved by using the functionality provided by the OpenMM 4.1 library and by writing extensions to the OpenMM 4.1 library where the required functionality was not available. In order to enable full integration of the two libraries, modifications were also made to the OpenFOAM code. Unlike the original CUDA-based hybrid solver, the final hybrid CPU-GPU solver used the OpenCL platform of OpenMM 4.1. Wherever possible, attempts were made to replicate the original OpenFOAM functionality in the GPU version as closely as possible.
In order to enable structured progress through the code development needed to complete this project, the work was divided into three main work packages. This section describes these work packages and the work done in detail.
WORK PACKAGE 1: OpenMM/OpenFOAM hybrid solver using the OpenMM-OpenCL platform
The very first version of the OpenFOAM-OpenMM hybrid Molecular Dynamics solver, with the pairwise force interactions calculated on the GPU, pre-dated this project and was implemented using the OpenMM-Cuda platform. However, at the time of initiating the present project the focus of the development work and support of the OpenMM team had shifted to the OpenCL platform due to its open and standardised nature and its applicability to multiple accelerator platforms and CPUs. Owing to this shift in strategy of the OpenMM team, we decided to replicate the Cuda solution in OpenCL, and then to implement the rest of the work proposed using the OpenMM-OpenCL platform.
Work package 1a: Incorporate the OpenMM-OpenCL platform into OpenFOAM
The changeover from using CUDA to using the OpenCL platform was thought to be possible through simply changing an API call. An OpenMM API call getPlatformByName was provided which made switching between target platforms straightforward. However, when coupled with OpenFOAM, unlike the CUDA platform which ran without problems, the OpenCL platform produced a Floating Point Signal Exception (SIGFPE). Further investigation revealed that the exception was raised by a NaN operation in the OpenCL code which was intercepted by the OpenFOAM SIGFPE exception class. It was eventually discovered that the fault lay in the OpenCL Context class which ran an accuracy check kernel in order to determine which platform it was running on and then to select the most appropriate set of mathematics functions to use in the simulation. The check consisted of performing a number of test calculations on the CPU, using the built-in CPU Math library, and then copying the resulting values to the GPU, running the same test calculations with the built-in GPU Math library functions, and then comparing the results to determine the floating-point error. As it was known that the code would be run on the GPU platform, this accuracy check was disabled and the appropriate Math library was selected explicitly. This removed the SIGFPE error problem and successfully integrated the OpenMM-OpenCL platform with the OpenFOAM front end.
Work package 1b: Custom Lennard-Jones parameters and the virial coefficient in OpenCL
The simulation-setup interface in OpenFOAM enables the configuration of Lennard-Jones interaction potentials with custom (for example, experimentally-obtained) cross-species potential parameters, rather than requiring the use of the Lorentz-Berthelot mixing rules to calculate them from single-species values. To replicate this functionality in OpenMM, the CustomNonbondedForce class was used. This class accepts as an input a string specifying the pairwise interaction-potential equation which is then parsed inside OpenMM, using the Lepton parser. The potential equation can be customised to represent a wide range of different interaction potentials. In the hybrid solver, the Lennard-Jones parameters were automatically extracted from the simulation that was set up through the OpenFOAM front end. An appropriate string was then created automatically which could be passed to the CustomNonbondedForce class in OpenMM.
This work package also dealt with the calculation of the virial tensor, used to measure pressure in Molecular Dynamics simulations. The computational complexity of the virial tensor calculation is of order N2, where N is the number of atoms in the simulation. Since the pairwise force calculation is also of order N2, in the CPU-based OpenFOAM the virial tensor calculation is implemented inside the pairwise force calculation step to avoid unnecessary and expensive duplication of computational effort. To achieve the same in OpenMM, the OpenCL force-calculation kernel had to be augmented with the virial calculation. As the force calculation had already been delegated to the GPU platform at this stage, it made sense to proceed with the calculation of the virial tensor next, in order to complete the transition of the force calculation step to the GPU platform. As the OpenMM library is atom-centred and OpenFOAM is molecule-centred, an additional kernel, calculating the centres of mass of every molecule, also had to be implemented. Once the kernels were suitably modified, the necessary API modifications were made to enable the resulting virial tensor to be communicated to the top layer of the OpenMM hierarchical software architecture. The requisite modifications to the OpenCLContext class and OpenCLNonbondedUtilities class were also made.
WORK PACKAGE 2: Port remaining MD simulation steps to OpenMM-OpenCL platform
Work Package 1 had taken care of delegating the pairwise force calculation and the associated virial tensor calculation to the OpenMM-OpenCL platform. The Molecular Dynamics algorithm consists of a pairwise force calculation, velocity and position integration and appropriate control and measurement steps (Figure 2(a)). At this stage, only the most expensive step in the MD simulation, the pairwise force calculation, was being performed on the GPU (in OpenMM) and the remaining steps on the CPU (in OpenFOAM), as shown in Figure 2(b). This required OpenFOAM and OpenMM to communicate data to each other at every time step of the simulation. Profiling the hybrid implementation at this stage highlighted that, in addition to the speedup already achieved by porting the force calculation to the GPU, around 50% of the computational time was now being spent in communicating the information between CPU (OpenFOAM) and GPU (OpenMM). It was thought that removing this communication would allow for another significant increase in computational efficiency (Figure 2(c)). The sub-packages in this work package each addressed the GPU port of one of the remaining steps of the Molecular Dynamics algorithm.
Work package 2a: Connect the OpenFOAM and OpenMM molecular representation. Given the amount of data exchange necessary between the two libraries, a careful analysis of the OpenFOAM data structures was done at this stage and it was decided that the best way to proceed would be to replicate the OpenFOAM data management style inside OpenMM and, in so doing, to make the OpenMM data management routines customisable through the OpenFOAM front end. As a result, the OpenFOAM design patterns (in particular, the abstract-factory design pattern) were adopted and incorporated in OpenMM. The details of the implementation are strongly linked to Work Packages 2c and 2d and are described in the appropriate sections of this report. Work package 2b: Verlet integration algorithm Work package 2c: Measurement tools A number of measurement tools were proposed; only a subset of these were eventually implemented. The main obstacle to the implementation of measurement tools was the size of the data gathered in the measurement process and its storage. The measurements could either be kept in the GPU memory until the end of the simulation and then returned to CPU for processing and storage, or communicated to CPU every write interval and stored to disk to save on memory space. As the GPU memory space is limited and best used to run larger simulations, a decision was made to limit the measurements that were implemented to those requiring less memory for storage and to communicate the data back to CPU at either every time step or every write interval, as appropriate. The measurements that were eventually implemented in OpenMM included the total number of molecules, number density, mass density, total mass, kinetic energy, potential energy, total energy and total momentum. Other measurements were considered, but were found to require downloading large arrays of data. This was found to be counterproductive due to the slow nature of the CPU-GPU communication. In terms of the detail of implementation, an abstract-factory design pattern was used in the OpenMM Low-Level API to create the skeleton for the measurement tools, and a factory design pattern was implemented at the API level. In order to add a new measurement tool, a subclass of the pure base class (called Measure) was created and the appropriate measurement functionality implemented in the form of OpenCL kernels. The measured properties were then communicated back to the OpenMM API layer through access functions of the MeasurementTools class. Appropriate access functions had to be written for each new measurement. Work package 2d: Control tools Due to the limited time available within this project, a priority list had to be established, and it was decided that the most important control tools to be implemented were the Berendsen thermostat and the external force controller. However, the design pattern which allows for addition of new control tools is fully in place and further tools can be added if and when they become necessary.
WORK PACKAGE 3: Additional functionality for complex engineering simulations on the GPU Work package 3a: Application of controllers to sub-domains and measurements in zones and bins The fine-grained control and measurement were implemented by appropriately modifying the abstract-factory design pattern, MeasurementTools and ControlTools classes, written as a part of Work Package 2, to measure and control inside spatially-defined bins. The bin coordinates were set up at the beginning of the simulation by the OpenFOAM front end and the measurement and control classes accepted the bin information as input parameters. Three new classes were created at the OpenMM Low-Level API: MeasurementInBins, derived from the abstract class Measure, as well as BerendsenInBins and ExternalForceInBins, both derived from the abstract class Control. The measurement tools implemented in Work Package 2 efficiently used the shared memory on the GPU to reduce memory-access times. When a similar strategy was attempted for the fine-grained measurement, it was found that the size of the shared memory (48 KB per streaming multi-processor on NVidia Tesla M2075) became too restrictive, particularly when the number of spatial bins used by the simulation increased. A decision was made to use the global memory to perform measurements in bins and this incurred the associated global-memory access time penalty when compared to the optimised shared-memory implementation of Work Package 2. Storing the measurement bin arrays in the global GPU memory also decreased the amount of memory available to the simulation and consequently, when fine-grained measurements were required, the size of the simulation (the number of atoms/molecules) decreased proportionally to the number of bins used. The measurements which are available in the fine-grained mode include total number of molecules, total mass of molecules, total momentum, kinetic energy and number of degrees of freedom. Tests on a GeForce GPU with 4GB GDDR5 showed that, using global memory to store the binned measurement information, up to 25 bins could be used. Work package 3b: Custom boundary conditions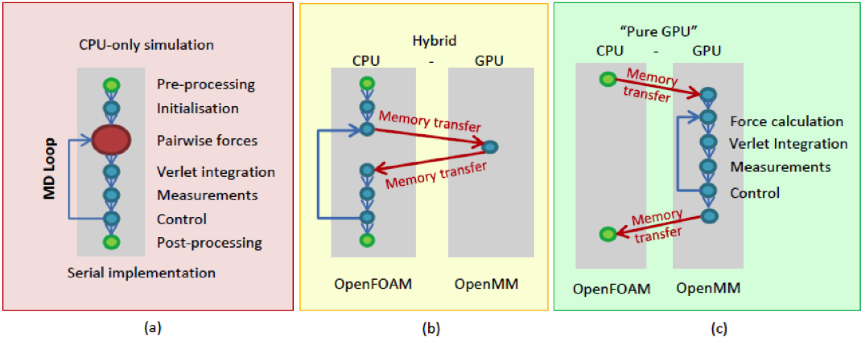
Figure 2: Transition from a pure-CPU to a pure-GPU MD solver
OpenFOAM contains a powerful set of algorithms for initialising Molecular Dynamics simulations in custom-defined simulation domains of arbitrary geometry. While the intention was for the main part of the MD simulation algorithm to be fully delegated to the OpenMM-OpenCL platform, OpenFOAM would remain the front end responsible for initialising the simulation and performing the necessary post-processing. OpenFOAM's data representation is molecule-based and OpenMM's is atom-based and thus a solution was needed to bridge this difference.
The next step in the Molecular Dynamics algorithm is the particle trajectory integration algorithm (see Figure 2). In OpenFOAM, the default integration algorithm is Velocity Verlet, while the OpenMM toolkit uses Leapfrog Verlet. As the intention was to make the GPU port fully compatible with the original OpenFOAM version, this work package implemented the Velocity Verlet algorithm in OpenMM. In the OpenMM API, a new subclass of the Integrator class was implemented and customised to follow the Velocity Verlet integration approach and the appropriate modifications were then made to the OpenMM-OpenCL kernels.
In Molecular Dynamics applied to fluids engineering problems, measuring various fluid-dynamic properties during the simulation run is commonplace and is a crucial research tool. As the focus of OpenMM was primarily on biological and biochemical simulations, most of the measurement tools that were required for applying Molecular Dynamics to fluid flow systems were not available and so had to be implemented.
OpenFOAM contains several control tools, all of which were proposed to be re-implemented for the GPU platform. The implementation of control tools followed a similar design pattern approach to that adopted for the measurement tools described in the previous section. The access functions for each of the measurement tools were implemented inside the ControlTools class in the OpenMM API layer.
The focus of the previous work packages was on porting the Molecular Dynamics algorithm to the GPU platform and adding the necessary control and measurement tools that were used to control and measure the average fluid-dynamic properties in the simulation domain as a whole. In Work Package 3, attention was turned to implementing additional functionality, which would enable more complex simulations to be run.
The aim of this work package was to provide a fine-grained measurement and control capability, necessary to enable control and measurement in sub-sections of the simulation domain. Fine-grained control enables complex simulation domains, for example, reservoirs connected with nanotubes, while fine-grained measurement enables a more detailed profiling of the fluid-dynamic properties during the simulation.
This work package was the last proposed work package in the work plan and it was intended to address the implementation of non-periodic boundary conditions that would have enabled simulations with complex boundaries. Due to time constraints, this part of the work programme was not implemented and is left as future work.
- Performance Evaluation
In order to provide an up-to-date appraisal of the difference in performance between the newly-developed hybrid solver and the CPU-based OpenFOAM library, OpenFOAM's performance has been re-tested to provide a new baseline for this report. This re-testing was necessary because, within the duration of this project, the MPI-based OpenFOAM also underwent significant revision to improve its performance.
The OpenFOAM version used in the James Weir Fluids Lab is 1.7.x. Although the official OpenFOAM version has moved on to version 2.2.1 at the time of writing this report, that version does not include the MD functionality developed inside the Lab. Porting this functionality from version 1.7.x to version 2.2.1 would require a major programming effort, but may indeed happen in the near future.
Table 1 shows the performance (time per MD timestep, in seconds) of the currently-used state-of-the-art pure-CPU OpenFOAM 1.7.x MD solver on a simple atomistic case without measurements and control tools.
| Number of cores → Simulation size ↓ |
12 | 24 | 36 | 48 | 60 | 72 | 96 |
| 119,164 | 0.394 | 0.361 | 0.225 | 0.196 | 0.157 | 0.115 | 0.085 |
| 238,328 | 0.706 | 0.653 | 0.457 | 0.351 | 0.267 | 0.208 | 0.153 |
Tests performed on NVidia Tesla M2075 (448 cores, 6GB GDDR5), available in ARCHIE-WeSt, showed that the choice of the OpenCL platform as the more optimised platform at the time was a sensible one. The hybrid solver at the end of Work Package 1, with force calculations performed on GPU, was around 2.2-times faster than the equivalent solver implemented using the OpenMM-CUDA platform, which was a precursor to this project (see Table 2). Due to circumstances beyond our control, it was not possible to use identical test cases for both tests. The simulation sizes shown in Table 2 differ slightly, with the test cases used for the new OpenCL-based solver being slightly larger than the ones used with the old CUDA-based solver. Table 2 also shows that, at this stage, the performance of the hybrid solver rivalled that of the MPI-OpenFOAM when run on approximately 58 CPU cores.
| Simulation size in atoms | Time per time-step in seconds using the original CUDA hybrid code | Simulation size in atoms | Time per time-step in seconds using the new OpenCL hybrid code |
| 118,638 | 0.35341 | 119,164 | 0.164 |
| 237,276 | 0.68253 | 238,328 | 0.2962 |
The port of the Verlet integration algorithm to the GPU platform (Work Package 2b) provided an additional speedup which meant that the new hybrid solver was then 3.3-times faster than the original CUDA solver (Table 3). The performance of the hybrid solver at this stage rivalled that of the MPI-OpenFOAM when run on approximately 79 CPU cores (cf. Table 1).
| Simulation size in atoms | Force calculation on the GPU (Work Package 1a) | Force calculation + Verlet integration on the GPU (Work Package 2b) |
| 119,164 | 0.164 | 0.106 |
The addition of measurements to the GPU platform (Work Package 2c) increased the computational time step slightly, as shown in Table 4. The time shown includes the time required to download the measurement data structure from GPU to CPU at every time step. As the measurements were then being performed on GPU, it was no longer necessary to download the molecular positions, velocities and forces to CPU at every time step. The hybrid solver was then equivalent to MPI-OpenFOAM when run on approximately 73 CPU cores.
| Simulation size in atoms | Force calculation + Verlet integration on the GPU (Work Package 2b) | Force calculation + Verlet integration + Measurements on the GPU (Work Package 2c) |
| 119,164 | 0.106 | 0.113 |
The addition of the control tools to the GPU platform (Work Package 2d) increased the computational time again, as shown in Table 5. The time reported includes the time necessary to perform control and measurement on GPU and the time required to download the measurement data structure to CPU every time step. The performance of the hybrid solver then compared to MPI-OpenFOAM when run on 71 CPU cores.
| Simulation size in atoms | Force calculation + Verlet integration + Measurements on the GPU (Work Package 2c) | Force calculation + Verlet integration + Measurements + Control Tools on the GPU (Work Package 2d) |
| 119,164 | 0.113 | 0.1151 |
Expanding the functionality of the measurement tools to apply in bins increased the computational time to that shown in Table 6. When compared to the implementation of the measurement tools across the entire simulation domain, the increase in computational time could be explained in terms of the overhead of assigning the calculated measurements to appropriate bins, the GPU memory management required to maintain a bin data structure and the download of the bin measurement array to CPU every time step. The number of bins used for this test case was 10 and the performance was comparable to MPI-OpenFOAM when run on 63 CPU cores.
| Simulation size in atoms | Force calculation + Verlet integration + Measurements on the GPU (Work Package 2c) | Force calculation + Verlet integration + Measurements in bins on the GPU (Work Package 3a) |
| 119,164 | 0.113 | 0.1449 |
The control tools were also extended to work in bins, however, coding issues prevented reliable test results for the Berendsen thermostat from being obtained at this stage and the related work is ongoing. As this information may still be relevant to the prospective user of the code, the computational time per time step for measurements computed in 10 bins, the external force controller applied in 10 bins and the Berendsen thermostat applied to the entire simulation domain is reported in Table 7 instead. As in the previous cases, the reported computational time includes the time taken to perform measurements in bins, control in bins and in the entire domain, and the time necessary to download the measurement data structure to CPU every time step. The performance is comparable to MPI-OpenFOAM when run on approximately 63 CPU cores.
| Simulation size in atoms | Force calculation + Verlet integration + Measurements in bins on the GPU (Work Package 3a, part1) | Force calculation + Verlet integration + Measurements in bins + Control in bins* on the GPU (Work Package 3a, part 2) |
| 119,164 | 0.1449 | 0.1454 |
The virial tensor calculation on the GPU was the subject of Work Package 1b, since its implementation fitted naturally with the force calculation stage. However, as the process of calculating the virial tensor is also an instance of applying a measurement tool, development of the virial persisted throughout the work programme. Despite inserting the virial calculation code into a GPU-optimised force calculation kernel, already available in OpenMM, the addition of the virial calculation resulted in a heavy computational load. The reason was the size of the virial tensor (3x3) which implied not only a large computational burden, but also significantly longer GPU-CPU communication times. Table 8 shows the increase in computational time per time step when calculating the virial tensor inside the force kernel on GPU. To highlight the computational complexity of this particular stage, the time reported includes that for performing the pairwise force calculation, Verlet integration and virial tensor calculation on the GPU, as well as the time required to download the virial tensor for all molecules at every time step. The computational performance compares to MPI-OpenFOAM when run on 63 CPU cores.
| Simulation size in atoms | Force calculation + Verlet integration (Work Package 2b) | Force calculation + Verlet integration + Virial measurement (Work Package 2b+1b) |
| 119,164 | 0.106 | 0.1455 |
Table 9 shows an overview of the computational time per time step measured for the new hybrid solver at its various stages of development. It can be seen that the hybrid solver with force calculation and Verlet integration run on a GPU rivals the performance of MPI-OpenFOAM when run on 79 ARCHIE-WeSt CPU cores. The addition of measurements and control increases the computational time, as expected. Performing the measurements in bins increases the computational time even further, bringing the hybrid-solver performance in line with MPI-OpenFOAM when run on 63 CPU cores. Finally, computing the virial tensor slows down the calculation to a stage where the hybrid solver consisting of only the core MD algorithm (pairwise force calculation and Verlet integration) and the virial calculations becomes equivalent to MPI-OpenFOAM when run on only 63 CPU cores. A graph of these values is shown in Figure 3.
| Simulation size in atoms | WP 1a | WP 2b | WP 2c | WP 2d | WP 3a measurements | WP 3a control* | WP 1b Virial |
| 119,164 | 0.164 | 0.106 | 0.113 | 0.1155 | 0.1449 | 0.1454 | 0.1455 |
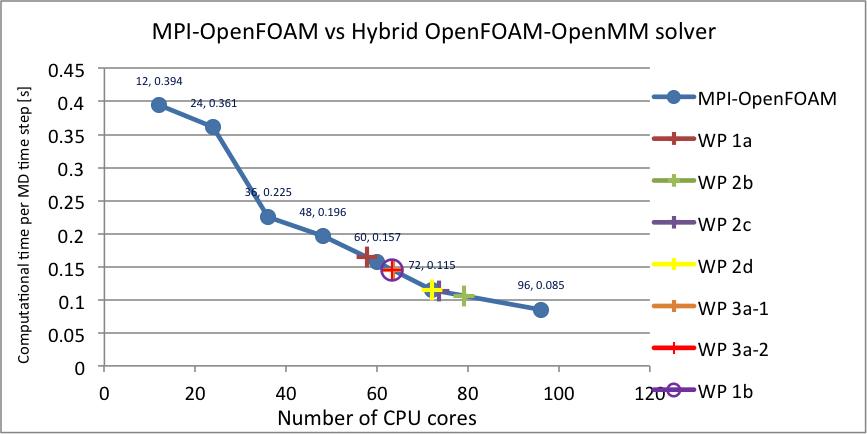
Figure 3: Comparison of the computational performance of MPI-OpenFOAM on varying number of ARCHIE-WeSt Intel Xeon X5650 2.66 GHz CPU cores and the hybrid OpenFOAM-OpenMM solver on ARCHIE-WeSt NVidia M2075.
- Discussion
The initial intention of the project described in this report was a straightforward port of an existing Molecular Dynamics algorithm available in pure-CPU OpenFOAM to a different platform, namely the GPU platform. As is always the case, however, many obstacles were encountered on the way and new areas for investigation were identified.
In the authors' view, the results shown in this report demonstrate the computational power of the GPU platform and highlight the fact that GPUs should be taken seriously as a co-processing tool for computational fluid dynamics at the nano-scale. Completed tests show that the main computational gain was achieved by porting the force calculations and the particle trajectory integration onto the GPU platform. The remaining work packages did not contribute a significant increase in efficiency, yet they did not increase the computational time either. Given further time, Work Packages 1b and 2c-3a could be revisited, allowing the relevant algorithms to be optimised further to better exploit the parallel nature of the GPU platform.
One of the main obstacles experienced during this project was the communication time required to download data from GPU to CPU on a regular basis. More could be achieved by hiding communication behind computation where possible, as well as by performing as much of the computation on the GPU as possible, thereby reducing the need to communicate with the CPU at every time step. This is not easily achievable though, as not communicating data to CPU means storing it in the (already limited) GPU memory instead, as well as putting GPU in charge of averaging, something that CPU is much better suited to. Investigating the best balance between the amount of computation done on GPU and that done on CPU would yield a useful separate project in its own right. It may well be that the GPUs are best suited to running the core MD algorithm efficiently and that any measurements would better be left to the post-processing stage.
The OpenMM version used and extended for the purposes of this project was version 4.1. The latest version of the official OpenMM library available at the time of writing this report is a highly-optimised version 5.1 which quite significantly outperforms the previous OpenMM versions. It is expected that incorporating the work described in this report into OpenMM 5.1 would provide an additional and significant computational speedup.
When comparing the performance of the hybrid solver to that of pure-CPU OpenFOAM, the CPU times were reported for only a very simple simulation case. Ideally, every stage of the hybrid solver should be compared to an equivalent simulation run in pure-CPU OpenFOAM through a strong-scaling experiment. Unfortunately, such insights could not be obtained, because the required number of ARCHIE-WeSt CPU cores was seldom readily available. In addition, the time required to run such thorough tests well exceeded the time that was available for the completion of the project.
At its best, the computational performance of the hybrid solver on one NVidia Tesla M2075 GPU, evaluated on an example test case, was comparable to MPI-OpenFOAM when run on 79 ARCHIE-WeSt CPU cores. Given that GPUs can be used as co-processors in desktop computers, this represents a significant increase in computational efficiency when compared to the typical number of CPU cores (8-12) available in such computers. In addition, the fact that high performance computers, like ARCHIE-WeSt, are a shared resource means that it is unlikely that 79 CPU cores will be regularly available to MD fluids engineers. It is much more likely that a single GPU node will be available consistently instead and, as this report demonstrates, one GPU node can provide a computational power equivalent to between 63 and 79 CPU cores, depending on the details of the simulated case. A further case in point is the cost of running simulations on high-performance computers, where the charge is levied per number of computational nodes occupied by the simulation. Replacing 6 CPU nodes (72 CPU cores) with 1 GPU node makes a noticeable difference to the simulation cost. Last, but not least, the advantage of running small-to-medium-size simulations on the GPU platform should not be overlooked, in particular, those simulations which are too small to be effectively decomposed on a large set of MPI cores because the communication slow-down is encountered early in the strong-scaling test. Such simulations may easily fit into the memory of a single GPU and are likely to run faster because the network communication is not required. In practice, the computational efficiency of the GPU code will always depend on the particular simulation case and a thorough understanding of the advantages and limitations of the GPU platform is of paramount importance if its computational power is to be exploited fully.
- End users
The project described in this report was implemented to increase the computational speed of Molecular Dynamics simulations used to conduct engineering research in molecular fluid dynamics. It directly benefits the activities of the researchers in the James Weir Fluids Lab, and other research groups or individuals with similar interests. Various research publications or presentations whose results use the code described in this report have already been, or are being, produced10-12.
- Licensing
OpenFOAM and OpenMM libraries are open source and developed under the GPL licence. In this spirit, the newly-developed source code will be made freely available. Currently, the code is undergoing further validation and testing, particularly in its interactions with OpenFOAM and stability for general release. The plan is to release it as an open source package on GitHub in late 2013, following this validation period. The authors are in regular communication with OpenCFD (ESI) Ltd, the OpenFOAM project architects, with a view to including this improved particle functionality into future releases of OpenFOAM. The authors are also communicating with the developers of OpenMM with the aim of introducing the additional functionality developed in this project into future standard releases of OpenMM. This is expected to come to fruition in 2014.
- Acknowledgements
This project was funded under the HECToR Distributed Computational Science and Engineering (CSE) Service operated by NAG Ltd. HECToR - A Research Councils UK High End Computing Service - is the UK's national supercomputing service, managed by EPSRC on behalf of the participating Research Councils. Its mission is to support capability science and engineering in UK academia. The HECToR supercomputers are managed by UoE HPCx Ltd and the CSE Support Service is provided by NAG Ltd. http://www.hector.ac.uk
The authors would like to thank Matthew Borg and Konstantinos Ritos of the James Weir Fluids Lab for their support and suggestions during the development.
- References
- James Weir Fluids Lab, http://cms.mecheng.strath.ac.uk/jwfluidslab/
- MPI, http://en.wikipedia.org/wiki/Message_Passing_Interface
- ARCHIE-WeSt, http://www.archie-west.ac.uk/
- OpenMM web page, https://simtk.org/home/openmm
- OpenFOAM web page, http://www.openfoam.org/
- OpenMM API manual web page, https://simtk.org/api_docs/openmm/api5_0/c++/
- OpenFOAM C++ source guide web page, http://www.openfoam.org/docs/cpp/
- P. Eastman, M. S. Friedrichs, J. D. Chodera, R. J. Radmer, C. M. Bruns, J. P. Ku, K. A. Beauchamp, T. J. Lane, L.-P. Wang, D. Shukla, T. Tye, M. Houston, T. Stich, C. Klein, M. R. Shirts, and V. S. Pande. "OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation." J. Chem. Theor. Comput. 9(1):461-469. (2013)
- P. Eastman and V.S. Pande. "Efficient Nonbonded Interactions for Molecular Dynamics on a Graphics Processing Unit." J. Comput. Chem. 31:1268-72. (2010)
- M. K. Borg, D. A. Lockerby, and J. M. Reese. "Fluid simulations with atomistic resolution: a hybrid multiscale method with field-wise coupling." submitted to J. Comput. Phys. (2013)
- M. K. Borg, D. A. Lockerby, and J. M. Reese. "Fluid flows in nano/micro network configurations: a multiscale molecular-continuum approach", 65th APS Fluid Dynamics Annual Meeting, San Diego, California. (2012)
- K. Ritos, M. K. Borg, S. Ivekovic, D. A. Lockerby, Y. H. Zhang, and J. M. Reese. "Nanotube water filtration membranes: A hybrid simulation approach", 26th Scottish Fluid Mechanics Meeting, Aberdeen. (2013)