Final
Report
Numerical Algorithms Group Ltd
Parallel Software Development Project
Uncertainty
Quantification using Differential Geometric Population MCMC
Ben
Calderhead∗1, Gary Macindoe2
and Mark Girolami†2
1CoMPLEX,
University College London, UK
2Department of Statistical Science, University College London, UK
9th
August 2013
Contents
1 Project Overview and Summary
2 Introduction
2.1
Motivating Scientific Examples
2.2 Benchmark Models
2.3 Cluster Configuration and Compiler Settings
2.4 Key Objectives
3 Work Package 1
3.1
Results
4 Work
Package 2
4.1
Results
5 Uptake and Dissemination of Software
6 Conclusions and
Future Work
*Gratefully acknowledges
support through the EPSRC “2020 Science” Fellowship Programme.
†Gratefully acknowledges
support through the EPSRC Established Career Research Fellowship EP/J016934/1
and a Royal Society Wolfson Research Merit Award.
1 Project Overview and Summary
Uncertainty
quantification is vital in many scientific areas. In particular, the Bayesian approach provides
a consistent inductive logic for reasoning about systems where measurements and
models contain often high degrees of uncertainty. A number of competing hypotheses
regarding the underlying structure of a system of interest can be encoded as
mathematical models, and we may use this Bayesian statistical framework to
decide systematically which model is best supported by the observed data and
may therefore be best employed for predictive purposes.
This project
developed an efficient C implementation of a parallel Differential Geometric
Markov Chain Monte Carlo (MCMC) sampler within a general population MCMC
framework using MPI. This state of
the art statistical methodology allows key Bayesian quantities to be accurately
estimated by drawing samples from a sequence of tempered probability
distributions bridging from the prior to the posterior. The posterior probability distribution
gives estimates of unknown parameter values in the statistical model given the
available data, and samples from the additional tempered distributions may be
used to estimate the marginal likelihood for comparing models.
The software
developed enables systematic comparison of model hypotheses for systems described
using coupled systems of nonlinear differential equations, such as gene
regulatory networks, and systems described using aggregated Markov processes,
such as ligand-gated ion channel mechanisms in the biological sciences. This
software can be deployed on any modelling problem where nonlinear differential
equations or Markov processes are used and as such has the potential to have a
widespread impact especially as the size and complexity of such models is
growing.
The C codes in
this project deliver scalable performance, which is up to 2 orders of magnitude
faster than the original Matlab implementation. Further, its modular design allows it to
be easily extended to enable statistical inference over additional classes of
models thus ensuring it widespread utility to practising
scientists and engineers.
2 Introduction
The quantification
of uncertainty is now being widely recognised as
essential to the enquiry and development process in the fields of science and
engineering1. Such
approaches drive a wide range of decision-making within industry, academia and
government, and there is consequently a very strong demand for software to
support this aim. In particular,
the use of Bayesian methodology for making predictions and quantifying
uncertainty both in data and models has massively increased over the last 15
years, in part due to the increased computing power available. Alongside this increase in computational
ability, advances in Bayesian algorithms also play a significant role in making
inference more feasible over complex statistical models.
The Bayesian
paradigm involves representing unknown variables or parameters of a statistical
model in terms of probability distributions, which are then updated according
to the rules of conditional probability (Bayes Rule) to take into account the
available data. This approach
provides a consistent inductive logic for reasoning about systems where
measurements and models often contain high degrees of uncertainty. The main scientific question that we wish
to answer may be posed as follows: Given a number of competing hypotheses
regarding the underlying structure of a system, how may we decide which
statistical model is best supported by the observed data and may therefore be
best employed for predictive purposes?
The Bayesian approach offers a systematic and quantitative way of
answering this, whilst avoiding overfitting to the data. However, such an
approach is computationally intensive; rather than calculating a single value
via optimisation, the answer instead takes the form
of a probability distribution, which must then be integrated to obtain the
required marginal likelihood for model comparison. For more realistic nonlinear models the
resulting densities and integrals are generally not analytic and must be
approximated.
Markov chain Monte
Carlo (MCMC) methods allow samples to be drawn from arbitrary probability
distributions and therefore provide a general approach to performing Bayesian
analysis for wide classes of statistical models. In this project we have produced
efficient parallelised C code which translates the original Matlab
implementation of state-of-the-art MCMC methodology based on incorporating the
Riemannian geometry induced by the statistical model of interest. This ``Differential Geometric MCMC''
methodology uses geometric information to explore the target distribution up to
2 orders of magnitude more efficiently than previous state-of-the-art MCMC
methods, particularly for high dimensional and strongly correlated densities. However, despite this dramatic
improvement in statistical efficiency the original Matlab code was still too
slow to allow realistically complex models to be analysed within a reasonable
timeframe.
This C code
delivers the capability of allowing such models to be analysed within a
Bayesian framework in hours rather than days by exploiting the speed-ups that
may be obtained through parallelisation on a High-Performance Computing Cluster
using MPI and OpenMP.
1 The NSF and SIAM in the USA are supporting national initiatives in formalising research in Uncertainty Quantification. Likewise the EPSRC in the UK has recently funded a large Programme grant on Uncertainty Quantification in engineering inverse problems led by Prof. A. Stuart, University of Warwick, of which M.A. Girolami is a co-investigator EP/K034154/1
2.1
Motivating Scientific Examples
We chose
particular scientific applications to be the motivating examples for driving
this software development; biochemical models described by nonlinear
differential equations and ligand-gated ion channel models described by
aggregated Markov processes. These
examples are based on current active research areas within Computational
Statistics (and collaborators) at UCL, and have the potential to make a large
impact within in the wider international research community. The software's framework has been
designed to be general enough to allow arbitrary models to be analysed by
specifying functions defining their likelihood and corresponding geometric
quantities. This allows the
computer code to be easily extendible to other statistical and scientific
applications in the physical sciences and engineering.
2.2
Benchmark Models
Two differential
equation models were used for benchmarking the improvement in speed over the
original Matlab code. The Fitzhugh-Nagumo model consists of a system of nonlinear differential
equations with 3 parameters and 2 initial conditions, resulting in a 5
dimensional probability distribution to be inferred. 1000 data points were generated from this
model by solving the system with parameters [0.2, 0.2, 3] and initial
conditions [-1, 1], and then adding Gaussian distributed perturbations to
simulate noise. The full details of
this model may be found in [1]. The
second nonlinear differential equation model describes a circadian biochemical
network in the plant Arabidopsis thaliana,
which consists of 22 parameters and 6 initial conditions and results in a 28
dimensional probability distribution to be inferred. 10 data points were generated for each
species (in order to simulate realistic experimental data) by solving the
system using the parameter values detailed in [2], where full details of the
model may also be found. In both
cases, the standard deviation of the noise was chosen to be 1 percent of the
standard deviation of the model output for each species.
We also considered
an ion channel model described by an aggregated Markov process. The Markov process used consists of 5
states with 10 parameters, the full details of which are available in [3]. A dataset consisting of 6700 data points
was generated to represent a realistically sized data set for the purpose of
benchmarking.
The settings for
the population MCMC sampler are described in the Sections 2 and 3, and the test
scripts and datasets are also included in the code release for reproduction of
results.
2.3
Cluster Configuration and Compiler Settings
All benchmarks
were performed on the Statistical Science Computer Cluster at UCL. This cluster consists of 52 nodes, each with
2 quad-core 2.7GHz Opteron AMD processors with 8GB of memory. The nodes are connected using 10Gbit ethernet and based around the open source linux distribution Rocks 5.3 (http://www.rocksclusters.org). Our software was compiled using gcc version 4.1.2 with LAPACK version 3.4.2, which includes
BLAS and CBLAS, and with the ODE solver package Sundials version 2.5.0. We also used OpenMPI
version 1.6 to enable parallelism on the cluster. The following flags were employed during
compilation:
-std=c99 -pedantic -Wall -Wextra
-march=opteron -02 -pipe
2.4
Key Objectives
The main
objectives for this project were as follows:
· Implementation of test
suite for MCMC update
functions.
· C implementation of MCMC
update functions using OpenMP.
· C implementation of population MCMC using
MPI.
· Matlab and R wrappers to run the developed
C code.
The first three of
these objectives were successfully completed resulting in a usable C
implementation that offers up to 2 orders of magnitude speed improvement over
the original Matlab code. The last
objective of writing wrappers to allow Matlab and R to directly run the C code
was not achieved due to time constraints, however this would be a useful
addition for future work. In the following
sections, an overview of each of these objectives is given in the order they
appear in the work packages. The
outcomes of the key objectives and resulting speed improvements compared with
the baseline Matlab code are also presented.
3
Work Package 1
The first task
involved refactoring the existing Matlab code and planning the structure of the
C implementation. The MCMC update
functions were then implemented in C using OpenMP to
accelerate the computation using multiple cores. It was decided not to implement the
Matlab wrappers at this stage, but to leave all interfaces to other software
packages until the end of Work Package 2, once the full sampler had been
implemented in C. This decision was
taken to ensure we would have time to deliver a fully working piece of software
coded entirely in C and took into account the reviewers' comments of our
original proposal, where it was mentioned that delivering the wrappers for
Matlab and R, in addition to the entire C implementation, would be rather ambitious. This proved to be a wise decision, as we
eventually ran out of time to deliver the wrappers, but did successfully
complete a fully working sampler in C, which we describe further in Work
Package 2.
Unit tests were
written to ensure correct outputs from the MCMC update functions were obtained
after being rewritten in C. In
particular, tests were written to confirm correct model outputs, likelihood
functions and geometric information required for the MCMC sampler, such as
gradients and metric tensors, for 100 randomly chosen inputs and the required
tolerance for passing each test was set to 1e-6. The test suite was fully automated in CUnit using random inputs and their corresponding outputs
generated from the original Matlab code.
3.1
Results
For this work
package, the C implementations of the MCMC update functions were compared
against the corresponding update functions in Matlab for each of the benchmark
models. The update functions
calculate the likelihood function of each model for a given set of parameters
and dataset, along with gradient and geometric quantities required for the
sampler. Timings were averaged over
1000 different parameter sets and performed on 4 cores using OpenMP enabled code.
These functions represent the main workload of a Monte Carlo sampler, as
they must be evaluated many hundreds of thousands of times.
|
Model |
Matlab
(sec) |
C
(sec) |
Speed
Improvement |
|
10 Parameter Ion Channel |
0.250678 |
0.003683 |
x68 |
|
5 Parameter ODE |
0.061808 |
0.001818 |
X33 |
|
28 Parameter ODE |
0.106800 |
0.019113 |
X6 |
4
Work Package 2
The main item of
work in this section was the development of the full population MCMC framework
in C using MPI, which utilises the MCMC update and likelihood
functions developed in Work Package 1.
The outputs of the software are samples drawn from a posterior
probability distribution, and summary statistics (mean and standard deviation)
of these outputs were checked against those obtained from the original Matlab
code to ensure correctness of the overall algorithm. This part of the algorithm was
structured in a modular form that allows additional ODE and aggregated Markov
models to be quickly added, as well as enabling easy integration of different classes
of models in the future.
An additional item
in this Work Package was the development of wrapper codes for accessing the
accelerated C code from Matlab and R.
Unfortunately this was not possible to achieve within the timeframe of
the project, however we were able to include the functionality that the
software output can be saved in a native Matlab and R-readable format for easy
post-processing of results.
4.1
Results
We first compared
the performance of our C code with the original Matlab code using the ion
channel model, where baseline performance is given by the Matlab code running
on a single core. We observed that
the C code offers much higher performance, as we might expect from the results
in Work Package 1, and exhibits better efficiency than Matlab when scaled up to
8 cores, as shown in Figure 1. Of
course the advantage of a C implementation is that we may use many more cores
for computation, without the need for expensive Matlab toolboxes to allow
this. Figure 2 demonstrates the
scaling efficiency up to 100 cores, where performance is 60 times better than
using a single core.
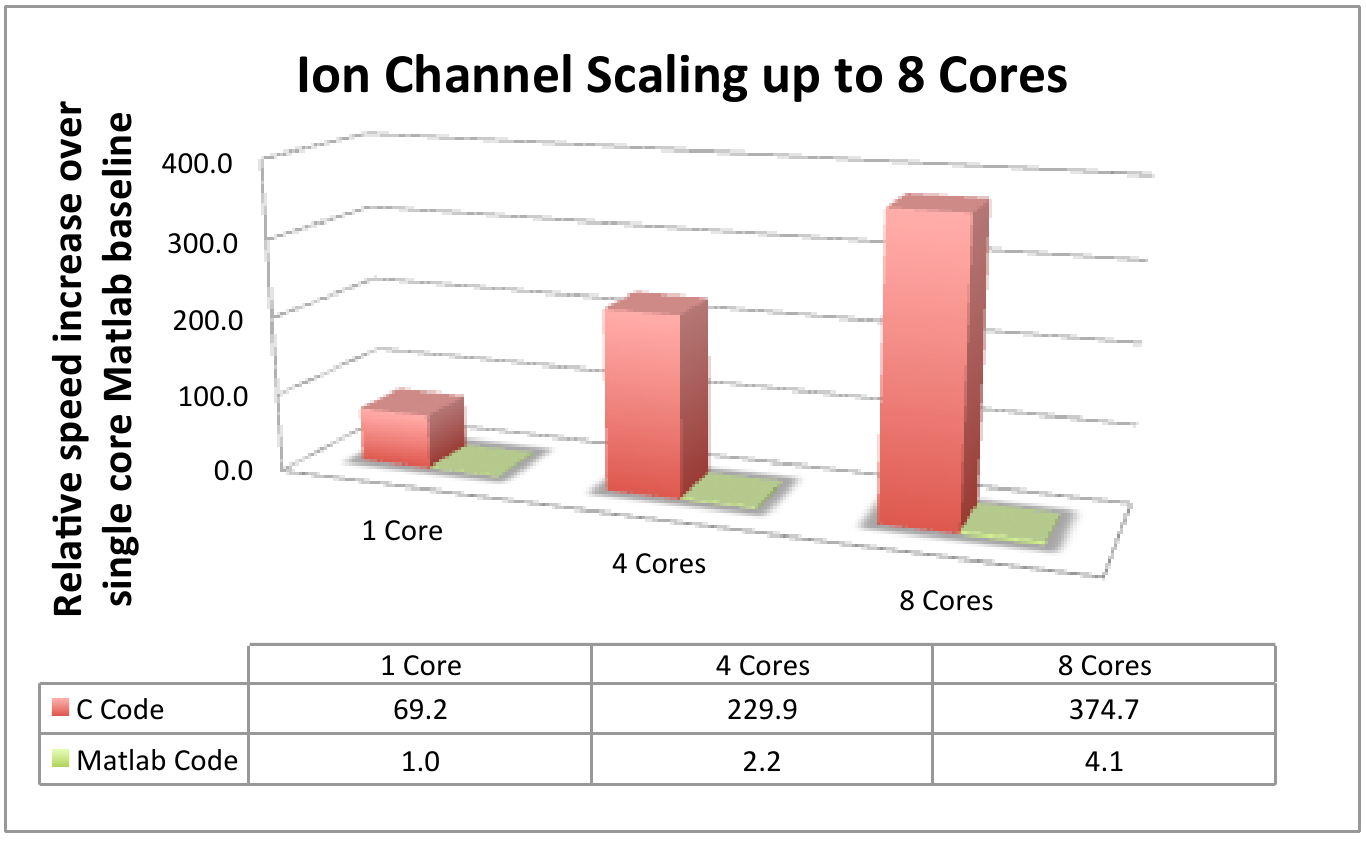
Figure 1: Comparative results using the ion channel
model with single core Matlab as the baseline performance measure.
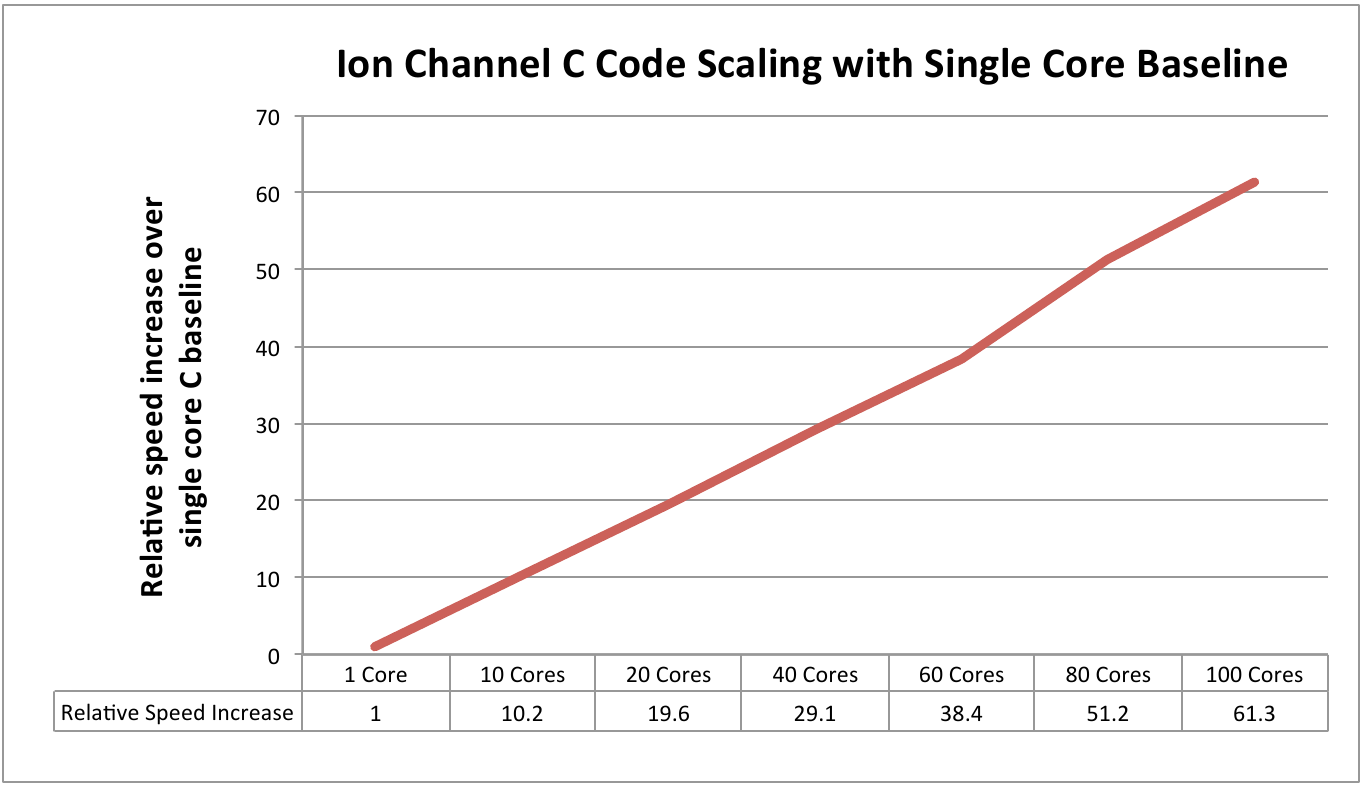
Figure 2: Scaling performance using the ion channel
model with single core C as the baseline measure.
We also compared
the performance of our C implementation using the two ODE model
benchmarks. Figure 3 shows the
scaling of the Fitzhugh Nagumo model with 5
parameters, compared to Matlab on up to 8 cores. Once again, above the increase in speed
due to the likelihood and geometry calculations, the performance of the C
implementation also scales more efficiently, as shown in Figure 4. Finally, we observed that the efficiency
of our largest ODE model with 28 parameters across increasing numbers of cores
scaled in a similar manner to the previous two example models, as may be seen
in Figure 5. In all cases the
communication overhead is perhaps fairly large relative to the individual
computation on each core, and so we might expect this performance to improve as
we consider larger models in future.
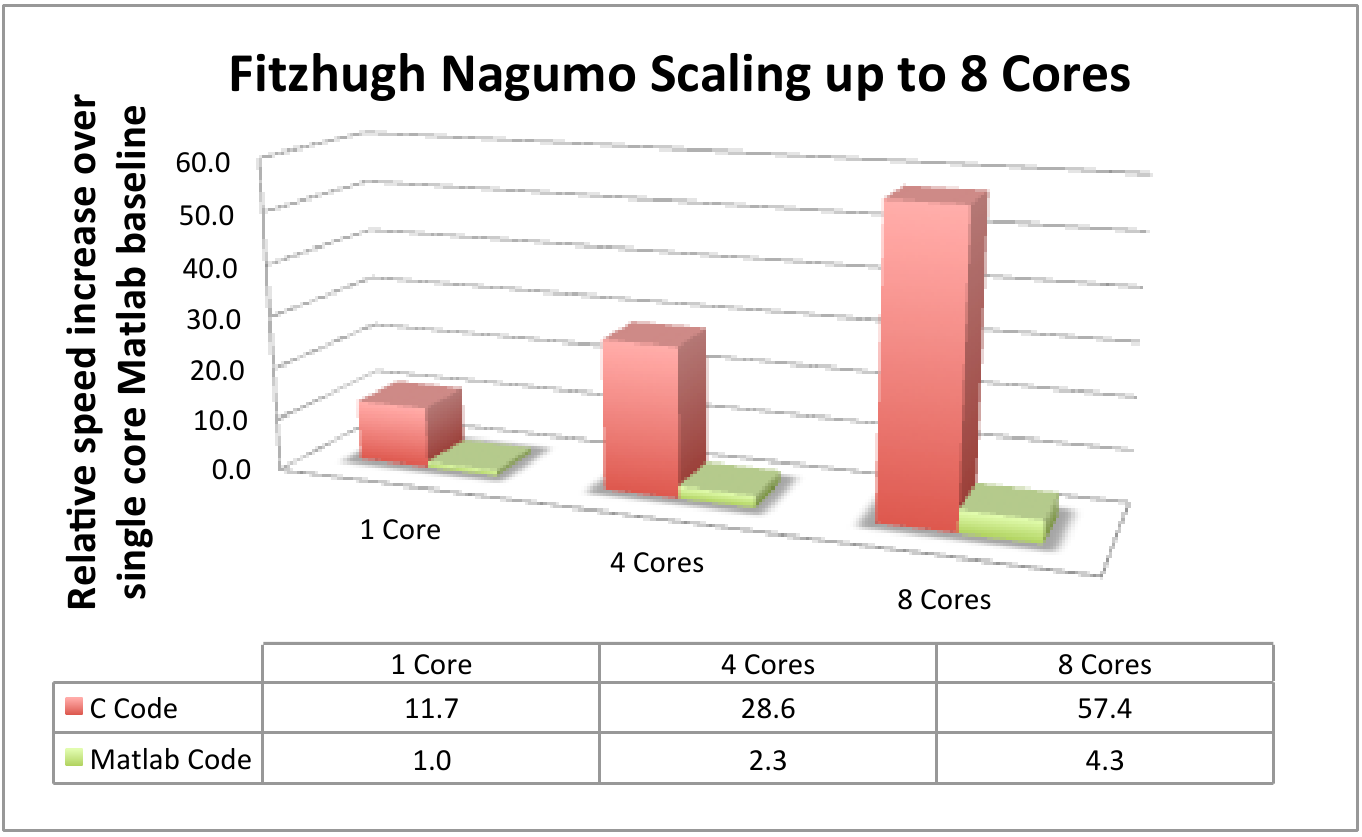
Figure 3: Comparative results using the Fitzhugh Nagumo model with single core Matlab as the baseline
performance measure.
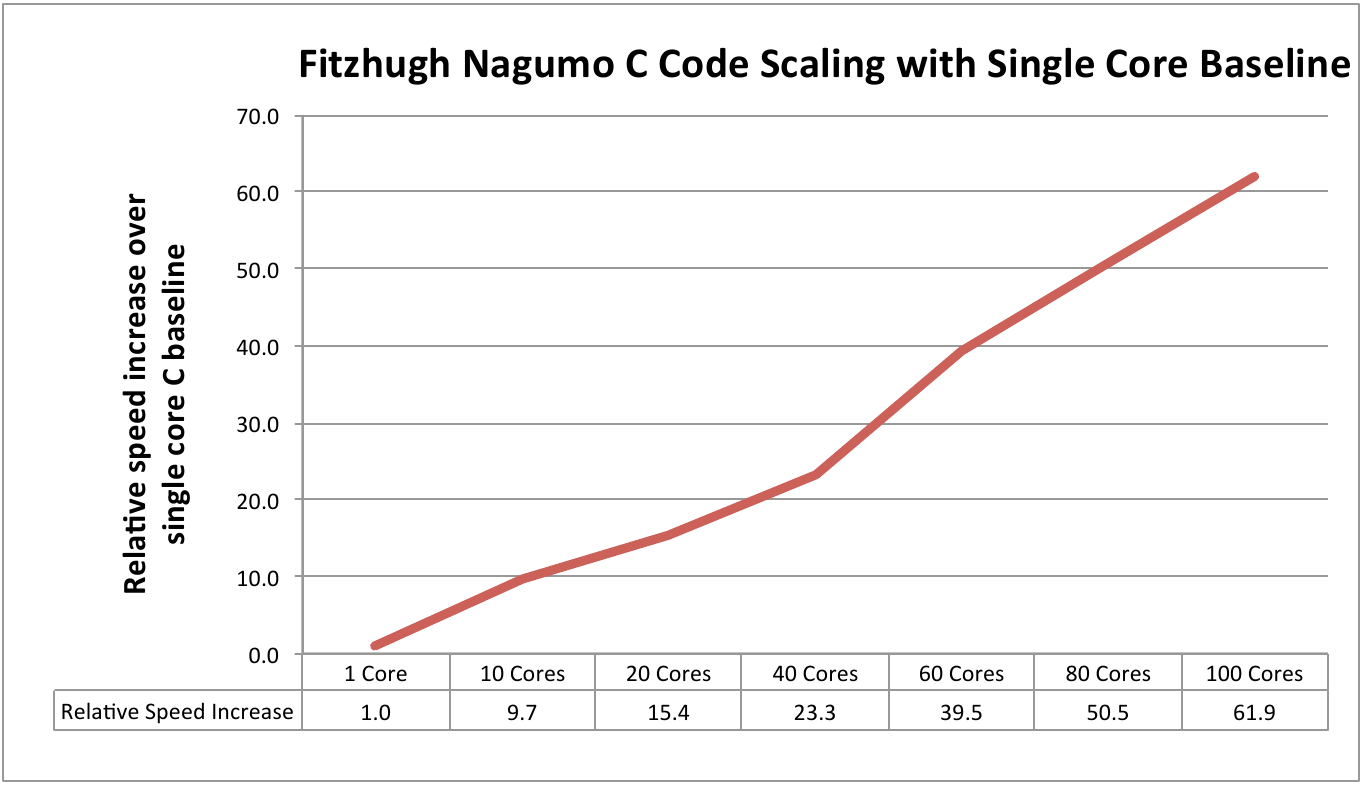
Figure 4: Scaling performance using the Fitzhugh Nagumo model with single core C as the baseline measure.
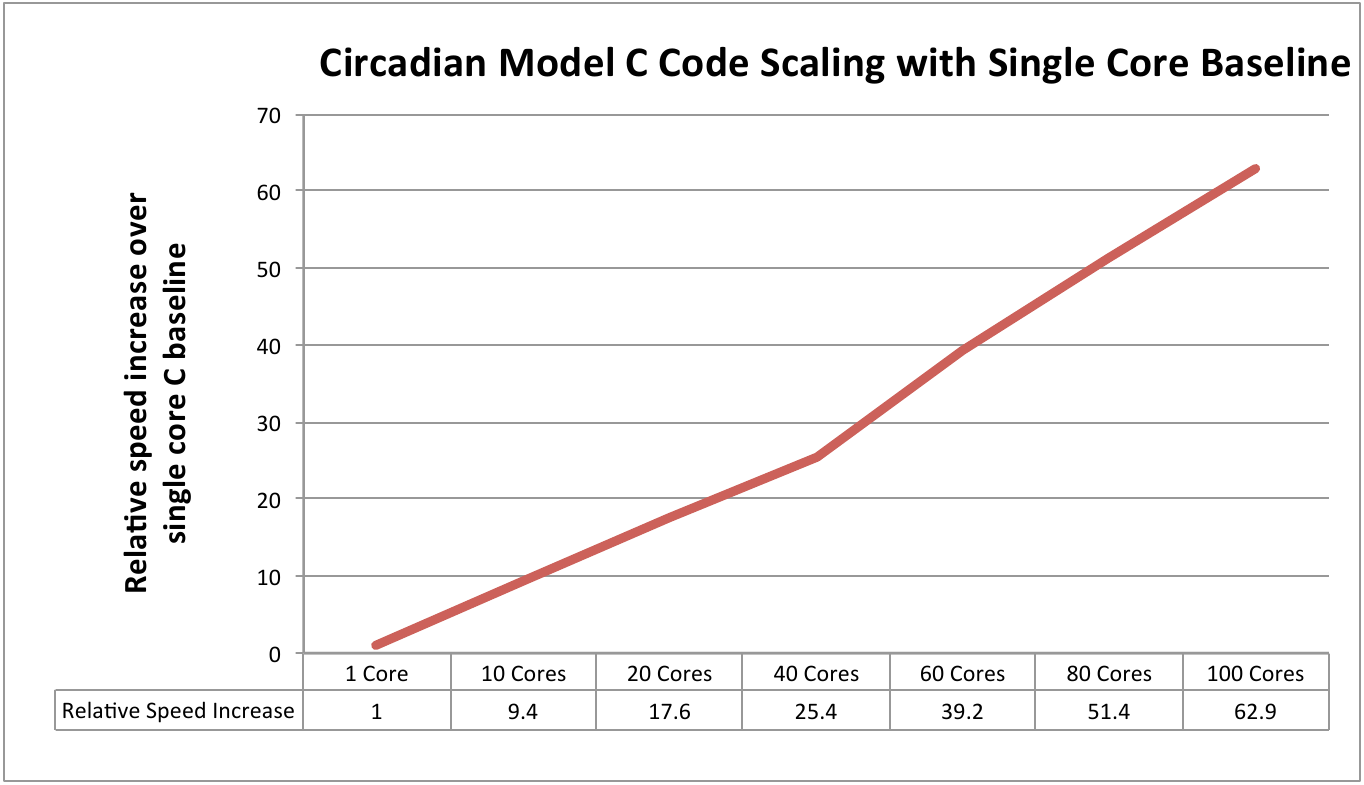
Figure 5: Scaling performance using the Circadian model with single core C as the baseline measure.
It is worth noting
that these results will have a real and immediate impact on increasing the rate
at which many of our motivating scientific questions may be answered; for
example, our C implementation can now generate 100,000 samples for the
Circadian model with 100 Markov chains in around 1 hour using 100 cores,
whereas the fastest previous Matlab version would take around 2 days with 8
cores. This offers a significant
advantage when there are large numbers of structural hypotheses to be tested
and compared.
5
Uptake and Dissemination of Software
The C
implementation we have developed is already being used in a number of projects
and collaborations involving Prof. Girolami and Dr. Calderhead. It is currently being used in a number
of work packages within ASSET (Analysing and Striking
the Sensitivities of Embryonal Tumours),
a 14 partner collaborative European project “tackling human diseases through
systems biology approaches”, see http://www.ucd.ie/sbi/asset/for
more details. The researchers
involved in this work include Prof. Mark Girolami (Statistical Science, UCL),
Prof. Heinrich Kovar (Children's Cancer Research
Institute, Vienna), and Prof. Walter Kolch (Systems
Biology Ireland).
This software is
also currently being used in a collaboration between
Prof. Girolami, Dr. Ben Calderhead (CoMPLEX and Statistical Science, UCL), Dr.
Lisa Hopcroft (Paul O'Gorman Leukaemia
Research Centre, Glasgow), and Prof. Tessa Holyoake (Paul O'Gorman Leukaemia Research Centre, Glasgow) to perform model
comparisons of hypothesised biochemical systems implicated in chronic myeloid leukaemia.
We will also be
presenting our software to a number of academic groups at universities in UCL,
Warwick, MIT and Heriot-Watt over the coming months, from whom
we obtained letters of support for our original application.
Further, we have
plans in place to bring this software and methodology to a wider audience
through publications in internationally leading journals. We are currently writing a paper for
submission to the Journal of Statistical
Software, as well as an application paper regarding differential equations
to be submitted to the biological journal Bioinformatics
and another application paper regarding ion channel models for submission to Nature Protocols. This will ensure the widest range of
exposure to both statisticians and biologists who may be interested in using
this software.
6
Conclusions and Future Work
We have
successfully developed a differential geometric population MCMC framework in C
and demonstrated the scalability and improvements in speed it offers compared
to the original Matlab code. In
particular, the software has enabled up to 2 orders of magnitude increase in
speed compared to the maximum 8 core implementation that was previously
possible in our original Matlab code.
This software is already having a significant impact in Europe-wide
projects and is expected to greatly increase the pace of scientific research in
the motivating scientific areas.
Future work will
involve developing interfaces to allow use of the C code from Matlab and R,
thus widening the audience.
This project was
funded under the HECToR Distributed Computational
Science and Engineering (CSE) Service operated by NAG Ltd. HECToR –
A Research Councils UK High End Computing Service - is the UK's national
supercomputing service, managed by EPSRC on behalf of the participating
Research Councils. Its mission is
to support capability science and engineering in UK academia. The HECToR supercomputers are managed by UoE
HPCx Ltd and the CSE Support Service is
provided by NAG Ltd. http://www.hector.ac.uk
References
[1] Riemann Manifold Langevin and Hamiltonian
Monte Carlo Methods, M. Girolami and B. Calderhead, Journal of the Royal
Statistical Society: Series B (with discussion) (2011), 73(2):123-214
[2] Statistical analysis of nonlinear dynamical
systems using differential geometric sampling methods, B. Calderhead and M.
Girolami, Journal of the Royal Society Interface Focus (2011), 1(6):821-835
[3] Bayesian Approaches for Mechanistic Ion
Channel Modeling, B. Calderhead, M. Epstein, L. Sivilotti
and M. Girolami, in “In Silico Systems Biology”,
Methods in Molecular Biology: Methods and Protocols, Springer