1. Introduction
DL_POLY_3 [[i]] is a general purpose package for classical molecular dynamics (MD) simulations developed by I.T. Todorov and W. Smith at STFC Daresbury Laboratory. The main purpose of this software is to enable the exploitation of large scale MD simulations on multi-processor platforms. DL_POLY_3 is fully self-contained and written in a modularised manner with communications handled by MPI. The rigorous and defensive programming style conforms to the Fortran95 standard (as checked by both NAGWare95 [[ii]] and FORCHECK95 [[iii]]) and guarantees exceptional portability. Parallelisation is based on equi-spatial domain decomposition distribution which guarantees excellent load balancing and full memory distribution provided the system’s particle density is fairly uniform across space [1]. This parallelisation strategy results in mostly point to point communication with very few global operations, and excellent scaling.
However, for a practical MD run excellent scaling of the CPU bound portion of the code is only one part of the total solution; it is also necessary both to save the results of the calculation to disk and to read the initial set up. As MD is a time-stepping algorithm the writing must be done periodically, and for a typical calculation it is performed every 100-1000 time-steps. As discussed in [[iv]] and [[v]] it is this reading and particularly the writing of data that has now become the bottleneck, especially at high processor counts and/or large problem sizes.
The work described in this report was carried out by Ian Bush who also gave a presentation on the project at the Cray User Group meeting which was held in Edinburgh 24-27 May 2010. The remainder of this report is organised as follows. In the next section we discuss in more detail the problem to be solved and earlier methods used by DL_POLY_3 and their drawbacks. In section 3, we shall describe a new method for writing that was released to users in version 3.10 of the code, and compare its performance with the earlier implementation. Section 4 discusses the new parallel reading method which will be in the next release of the code, and again we compare the performance with the previous methodology. Section 5 describes recent developments based upon netCDF [7], and finally we discuss how the improvements in I/O have improved the scaling of the whole code.
2. I/O in Domain Decomposed MD
The main I/O in DL_POLY_3 is, as is in most classical MD codes, the reading and writing of configurations representing frames of the time evolution of the modelled system. In both cases these are simply lists of the coordinates of the particles in the system, and optionally the velocities and forces acting on those particles. In DL_POLY_3 this has traditionally been performed using formatted I/O for portability. This is because while the MD run itself may be done on HPC facility or commodity cluster, the analysis of the results is often done on a workstation at the user’s own establishment. Thus the output resembles the following
Na+ 1
-184.5476802 -167.5198486 -185.5358832
-2.881253622 -4.727721670 2.235042811
-10840.17697 -5695.571326 -2740.726482
repeated many times, where the four lines are the atom's identity, position, velocity and the force acting upon it.
Although one may think this looks simple, there is one major complication as a consequence of domain decomposition scheme: As a parallel MD run progresses the original internal ordering of the atoms is scrambled, and furthermore atoms can migrate from one domain to another. Thus while the second atom as input may originally reside on processor 0 as the run proceeds it may migrate through the regions of space assigned to a number of other processors. The nett result is that while at the start of the calculation a particle is read in as the nth atom, at the end it may no longer be natural to write it to the equivalent place in the output file. One might contrast this with methods based upon solving differential equations on a grid. Despite both methods being domain decomposed in the latter case there is not usually this complication as the grid is fixed in space throughout the run.
Of course for the MD itself this is not an issue; obviously any numerical labelling of those atoms on input or output can make no difference to the results of the simulation. However, many post-processing software tools rely on the order of the atoms in the output file(s) to be maintained. One example of such is visualisation software that displays the time evolution of the positions of the atoms in the run. Such software must be able to track a given atom from one frame to another, and this is typically done by assuming that the position of a given atom within the output is conserved.
Thus for the user it is extremely convenient that the order of the atoms be conserved. On the other hand for the parallel application it is extremely inconvenient! It implies that a reordering of the data must be performed every time a configuration is written, an operation that can be expensive for distributed data applications. This reorganisation can be achieved in one of two ways (or a combination of both)
1. Between the CPUs: The CPUs communicate between each other to restore the original ordering of the data.
2. On the disk: By examination of the local data each CPU identifies where in the file the records for a given atom should be written, and simply writes directly into that section of the file.
Historically option 2 has been used by DL_POLY_3. This is made relatively simple by use of Fortran’s direct access I/O facility, especially given the very regular format of the output files. This is described more fully in [[vi]]. Two methods were implemented and tested:
1. SWRITE: Each processor in turn sends it data to processor zero, which deals with all the I/O by use of a direct access file.
2. PWRITE: Each processor writes its local data to the appropriate records in the direct access file.
As described in [[vii]] both these have their drawbacks. SWRITE is obviously not a scalable solution, and further the available disk bandwidth on modern parallel file systems often cannot be saturated by a single processor. On the other hand while PWRITE may scale with processor count the limited disk bandwidth may cause contention at the disk, and this is normally the case for large numbers of processors. However, this is not the only drawback of the PWRITE method. The Fortran standard describes the behaviour of a serial code. Therefore quite what should occur when multiple processes access one direct access file is not well defined, and in practice it is observed that on the Cray XT3/4 series using Lustre “spurious” NULL characters are introduced into the file when the PWRITE method is used. This problem is ultimately due to incoherence between the disk and caches.
Over and above the portability concerns the performance of the PWRITE method was simply insufficient to permit good scaling of the whole code, as described in [[viii]]. Therefore since [7] two new portable and potentially scalable methods have been developed, MWRITE and MWRITE_SORTED and they are the main focus for this paper.
However for very large systems there are two further problems. Though writing is much more important than reading, since the atomic configuration has to be dumped periodically throughout the run while reading is done but once, at large system sizes and processor counts reading the initial configuration can become prohibitively expensive. Further for large systems the formatted input and output files can become extremely large. We discuss both these points; a parallel reading method has been developed and is described, and an initial implementation of a netCDF [[ix]] version of the main I/O routines has also very recently been completed.
3. Writing Configurations
One solution to the portability problems associated with the PWRITE method is to use MPI-IO. If one wishes to preserve the format of Fortran Direct Access files, as is the case here for backward compatibility, this is particularly simple. In such files each record is the same length. A record can therefore be mapped onto a MPI derived type composed of an array of characters of the appropriate size. One then simply uses MPI_FILE_SET_VIEW to set the elementary data type for the file to be this data type, and subsequently all offsets within the file are calculated in units of the size of this type. The nett result is that MPI_FILE_WRITE_AT can be used exactly as for a Fortran direct access write except that
1. The program has to address the conversion of data to its character representation. For instance floating point numbers need to be converted to a string which is identical to the representation of the number obtained when using formatted output. In Fortran this is most easily achieved by the use of internal writes.
2. The indexing of the records is from 0, rather than from 1.
We are very grateful to David Tanqueray of Cray who brought this idea to our attention. He also kindly provided us with an implementation which we shall call MWRITE, and until version 3.09 of DL_POLY_3 this was the default writing method.
MWRITE is portable. However, as described in [8] and [[x]] its performance is not sufficient to enable good scaling of the whole code. This poor performance is for two primary reasons
1. Each I/O transaction is the data for just 1 atom, and is therefore very short (292 bytes).
2. With all the processors writing contention may occur at the disk, resulting in reduced performance.
To address these problems, the MWRITE_SORTED method has been developed. A very early prototype of this which lacked a number of facilities is discussed in [[xi]]. Here we discuss the full implementation as released to users. The ideas behind this are two-fold:
1. To avoid disk contention the data is gathered onto a subset of the processes within the job. These, the I/O processors, then perform the writing.
2. Within this subset before writing the data is sorted so that the particles are put back into their original order. As the data is now in the correct order this allows the writing of the data associated with many particles to be performed at once, thus allowing larger transactions and hence better I/O performance.
After these two stages the output itself is performed using MPI-I/O.
To avoid large memory overheads the gathering is done in batches. For instance consider a simulation of N particles with PI/O I/O processors. Rather than gather all the data onto the I/O processors, which may not be possible due to memory constraints, initially the data corresponding to the first PI/O*NB atoms is gathered onto the I/O processors, across which the atoms are evenly distributed, and then written out. Here NB, the batch size, is a parameter to be chosen. Then the next PI/O*NB atoms are gathered and written, and so on until all the data is written. This is simple to achieve as all that is required to efficiently identify the member of a given batch on a given core is a local sort of the atoms into their original increasing order. NB should be chosen according to two competing criteria:
1. It should be large enough to ensure that both the message passing in the gathering and the also the actual writing of the data to disk is efficient.
2. It should be small so that the memory overheads on the I/O processors are acceptable.
In practice the ranks of the I/O processors in the code are evenly spread throughout MPI_COMM_WORLD. It is our experience that on many systems the cores with the first n ranks reside on the first n-way multi-core processor, the next n on the next processor and so on. If this is the case the memory overhead is on the per-node basis, rather than per-core, and thus larger batching may be used as there is more available memory. However, it must be stressed that this method does not ensure this placement as there is no portable method within MPI to identify cores on the same or different nodes. The method will always successfully write the data to disk, though, assuming sufficient memory is available. MWRITE_SORTED has now been fully implemented, and is now released to the users as the default output method in version 3.10 of DL_POLY_3.
Figure 1 compares the performance of DL_POLY_3 versions 3.09 and 3.10 on phase2a HECToR [[xii]], a Cray XT4 system based on 2.3 GHz quad core AMD Opteron processors and a Lustre file system [[xiii]]. Each case was run 5 times, and the best time was chosen.
The physical system being studied is 216,000 ions of Sodium Chloride. The run is for 1000 time steps, and then the coordinates are written out once at the end of the job. This has been chosen as it is representative of the size of system that a number of groups are studying.
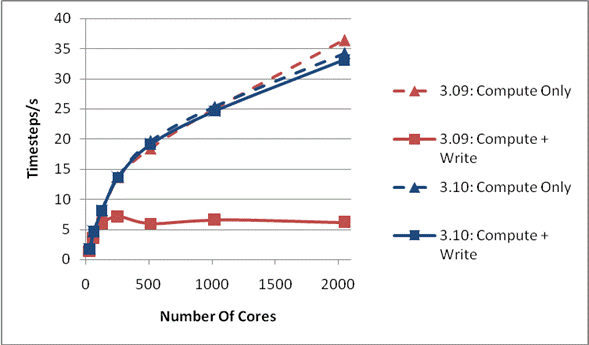
Figure 1: Write Performance for 216000 Ions
It can be seen from the compute only curves that the code is scaling well and that there is little difference between the versions. As the compute part of the code differs little between the two versions this is unsurprising. However, it can be seen that once the time for writing the final configuration is included the scaling is markedly different for the two code versions. Version 3.09 scales up to about 128 cores, and then the I/O inhibits any further improvement in performance, even though the compute is scaling well. The inclusion of the I/O time for 3.10, however, hardly makes any difference in the overall performance on the processor counts studied here, thus indicating a much improved I/O performance due to the new MWRITE_SORTED method.
In the above the default settings were used throughout for both the code and any environment variables that affect the behaviour of the Lustre file system. We feel that this is important as the majority of users will be unaware of what mechanisms can be used to tune the I/O performance of the code, and so will run with the defaults.
For the code the defaults have been chosen by examining how they affect performance on both HECToR and the earlier HPCx system [[xiv]]. It has been found empirically that a reasonable set of defaults for writing are
· 8P1/2 I/O processors, where P is the total number of cores.
· Batch the atoms 50,000 at a time.
· Write 5,000 the data associated with atoms per I/O transaction.
The reason for the last is the need to convert the numerical data into its character representation. Again a large number favours good I/O performance, but its range is limited by memory overheads. Of course it would be best if the code automatically optimised the side of the various buffers, but given the lazy allocation mechanism currently implemented in the kernel of the XT4 this is difficult to do, as it is difficult to identify how much unused memory is available.
More generally MWRITE_SORTED has been tested against the 34 DL_POLY_3 test cases which are available at [[xv]], and it has been found that on 256 cores of HECToR the writing in 3.10 is on average over 50 times quicker than in 3.09.
Table 1 shows the effective performance of the I/O for the two versions of DL_POLY_3 in terms of time and MByte/s. The time used to evaluate the performance includes all message passing and compute associated with the methods, hence the use of “effective” above. We choose this as it is this time that is of interest for the MD practitioner and programmer, not just the time to access the disks.
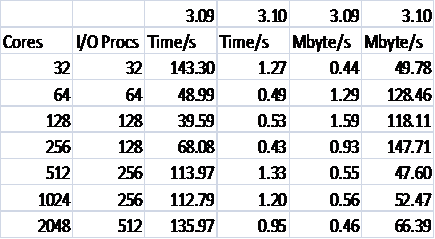
Table 1: Writing Performance for 216000 Ions
Firstly, it should be noted that despite running each case a number of times and taking the best result there is still appreciable noise in the data. Despite this a number of conclusions can be reached. Most importantly, it can be seen how much superior MWRITE_SORTED as implemented in version 3.10 is to MWRITE. It can also be seen that the default choices for the code result in the best performance at 256 cores. As this is the point to which the compute scales almost linearly (as seen in Figure 1) for this particular case this is indeed the place where we need the performance to be best as this is the number of cores that we would recommend the user to run it on to best make use of their computational budget. As such the degradation at higher core counts is not important, especially because, as can be seen for Figure 1, the overall effect on the code is minimal.
We have also tested whether MWRITE_SORTED scales sufficiently well with system size to allow larger physical problems to be studied on larger numbers of processors. This is illustrated in Figure 2. The system is again NaCl, but now the total size is 1,728,000 ions. Again, the system was run for 1000 time steps and a single configuration was dumped, and again the default I/O parameters have been used. It can be seen that the whole run, including I/O, is now scaling excellently to beyond 1000 cores. Also the peak writing bandwidth is much superior to that found for the smaller case at 810 MByte/s. We have not run this system using the 3.09 code as the time required for I/O makes it impractical.
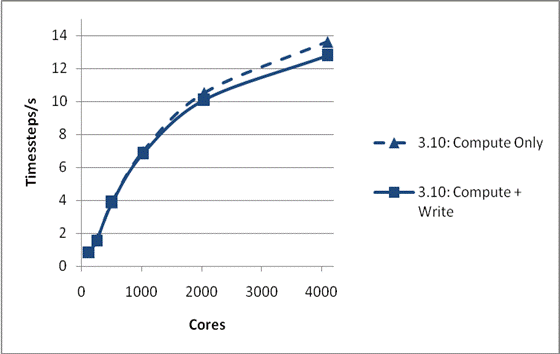
Figure 2: Write Performance for 1728000 ions
It is worth noting that larger system was generated from the original by use of DL_POLY_3’s nfold option. This allows a system to be scaled in directions parallel to each of the lattice vectors, and in this case the system was doubled in each of the x, y and z directions. To perform this requires a new configuration to be written. Version 3.10, using MWRITE_SORTED, completes this operation on 256 cores in 1.5 seconds. Version 3.09, using MWRITE, takes around 1 hour!
4. Reading Configurations
The currently released version of DL_POLY_3, 3.10, does not
read configurations in parallel. The method used is
· The core with rank zero in MPI_COMM_WORLD reads in a batch of the atomic data.
· Core zero then broadcast all the data for a given batch to all the other processors.
· From the batch a given processor extracts which atoms are in the domain for which it is responsible.
· Core zero reads in the next batch, and the above is repeated until all the data has been read from the file.
Reading the input data in parallel is both easier and harder
than writing. For reading no sorting is
required, and so as illustrated in the above scheme reading in large batches is
straightforward. However on writing the
programmer may specify the format so as to best suit has own needs, provided it
is within the constraints described in the DL_POLY_3 manual [[xvi]]. On the other hand for reading it is the user
that provides the file, and thus the program must be flexible enough to adjust
to any choices the user may make for his or her convenience. In practice, there are two important cases:
1. The input file was originally generated by DL_POLY_3 (e.g. for a restart or an analysis of a trajectory). In this case the format is exactly that chosen by the code.
2. The input file was generated by some other method.
In practice case 1 is both the more important and the easier to deal with. Therefore the new code automatically detects which category the provided input file falls into, and if it is case 1 it implements an optimised method, while if it is an example of case 2 a more general method is used.
The idea behind the two methods is essentially the same:
· A subset of the processors, again termed the I/O processors, each read in a batch of atomic data.
· Each I/O processor determines upon which core each of the atoms within its batch should reside.
· The I/O processors scatter their batch to the other cores so that each atom is received by the appropriate core.
· The I/O processors each read in another batch, and the process is repeated until all the data has been read from the file.
The main optimisation available if the format is known a priori (case 1 above) is that the length of each of the records in the input file is known. This permits
· Knowing a priori where each batch starts in the file. If this is not known in Fortran it is necessary to skip over each batch until the correct starting place is found, thus incurring an overhead.
· Optimal reading of each record, as the record length is known.
For either variants of the parallel reading method the potential benefits are that the reading is now in parallel and that the use of large I/O transaction lengths is retained from the original.
Table 2 shows the performance of the reading in version 3.10 and the new code. The example is the smaller NaCl case outlined above, and the format of the file allows the optimised method to be used.
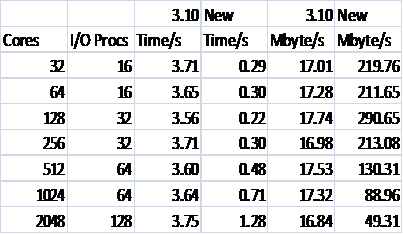
Table 2: Reading performance for 216000 Ions
Again all defaults for both the code, in terms of batch sizes and number of I/O processors, and the Lustre file system have been used. It can be seen that though there is a significant gain due to reading in parallel it is not as pronounced as for writing. This is unsurprising as in DL_POLY_3.10 though the reading is not done in parallel at least it uses large I/O transactions. The serial nature of the reading for the released 3.10 version of the code is also very clear, as is the fact that it is disk accesses that dominate the time, not the broadcasting of the batches. It can also be seen that the reading performance degrades more sharply at higher processor counts than for writing. This requires more investigation, and may indicate that the defaults for I/O used by the code can be optimised further. Currently, they are
· 2P1/2 I/O processors, where P is the total number of cores.
· Batch the atoms 50,000 at a time.
· Write the data associated with 5000 atoms at each I/O transaction.
Table 3 shows the scaling of the I/O performance for the larger case.
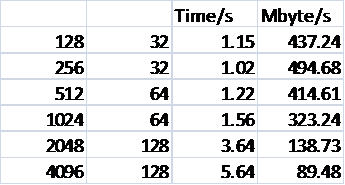
Table 3: Reading Performance for 1728000 Ions
Only data for the new method is shown. It can be seen that for this larger case a better peak I/O effective bandwidth is attained, and but that again there is a rapid fall off in performance.
5. NetCDF
In all released versions of DL_POLY to date the input and output files have always been readable by standard Fortran formatted read statements. For very large systems the reading and generation of such files may be undesirable for two reasons:
1. The conversion of the character representation of a number to/from its internal binary representation may impose a large overhead on the I/O performance.
2. Formatted files are usually appreciably bigger than binary files containing the same data.
In an attempt to deal with both these problems whilst retaining the portability of the input and output files very recently DL_POLY_3’s I/O subsystem has been extended to enable parallel reading and writing of netCDF [[xvii]] files. The chosen format for these files is based very closely on the published Amber convention [[xviii]], the major differences being
1. The files are opened as netCDF4 files so as to be able to use the parallel I/O capabilities in netCDF version 4 [9] provided by the use of HDF5 [[xix]].
2. Double precision is used to store the coordinates and velocities of the atoms, rather than single precision.
3. The forces are also stored, as per the formatted files in DL_POLY_3.
The resulting files are roughly one third the size of their formatted equivalent. Given that a trajectory history can result in files many hundreds of gigabytes long this is a very substantial saving.
The current implementation is based upon the earlier parallel I/O methods. All the sorting and batching is performed exactly as before, only the initialisation of the files, their closing and the actual I/O statements are changed. No effort has been made to optimise the various I/O parameters for the netCDF method, and those employed are exactly the same as those for the corresponding MPI-I/O based method.
Table 4 shows the results for writing for the smaller NaCl test case. It can be seen that the performance is very disappointing, especially when compared with that achieved via MPI-I/O upon which netCDF is itself ultimately based. The behaviour of the reading is similar. It is clear that further investigation is required in this area, for the potential benefits due to the savings in file size are very great.
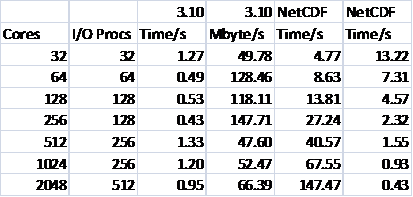
Table 4: NetCDF Writing Performance
5. Conclusions
Figure 3 shows the scaling of the latest version of the code with all the optimisation for a complete run, i.e. including all I/O, for the smaller NaCl case. Also shown is the scaling of the code released as version 3.09. It is clear that for systems of this size that the I/O problems that previously prevented scaling to large core counts are now solved.
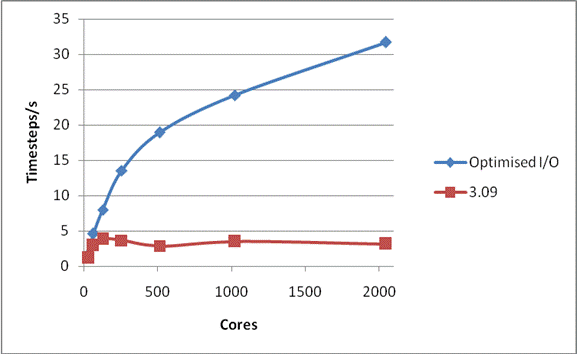
Figure 3: Total Performance for 216,000 Ions
Figure 4 shows the larger case. Only the newer code has been run. Again, the scaling is good even when I/O is included in the times.
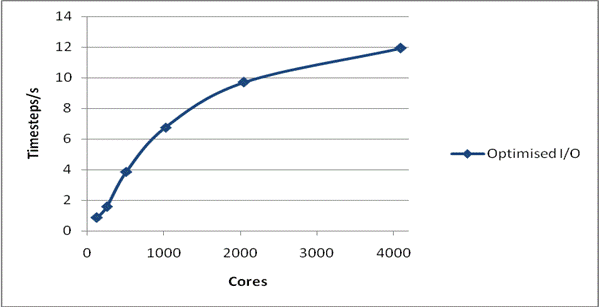
Figure 4: Total Performance for 1,728,000 Ions
The excellent scaling of the new code is
despite it passing many more messages than the old, especially at the
configuration writing stage. This is
because all the extra messages allow much better utilisation of the disks, and
slow though the network may be when compared to the computational power of the
CPUs, it is much quicker than the quickest of disks. Thus because of the slowness of I/O it is
worth a large scale reorganization of the data to obtain the best I/O
performance possible. That one should
optimise to get the best out of the slowest part of the problem is obvious,
this being the basis of virtually optimisation work. However the improvements
are not always as dramatic as in this case!
From the above it is clear that given a
suitable set of parameters DL_POLY_3 can now scale to thousands of processers
even when the I/O is included. This is
absolutely vital, for without the I/O portion of the code no science can be
performed, and the simulation simply becomes a synthetic benchmark will little
practical application. Now, however,
DL_POLY_3 users can perform real scientific runs on thousands of processors.
Acknowledgments
As mentioned above the authors would like to thank David Tanqueray for his insight that led to the MWRITE method, and his implementation of it. Though not the final version, without it none of the rest of the work could have occurred.
The authors would also like to acknowledge the
use of both the HPCx service and HECToR
– a Research Councils UK High End Computing Service, upon which the much of
this work was performed.
Ian Bush would like to
acknowledge the dCSE award from EPSRC that enabled much of this work.
About the Authors
Dr. Ian J. Bush has a BA in Chemistry (1989,
University of Oxford) and a PhD in Computational Chemistry (1993, University of
Oxford). He is a Senior Technical
Consultant at NAG Ltd. with particular responsibility for HPC applications in
the Computational Chemistry area; he is an author of CRYSTAL and has
contributed to DL_POLY_3 in a number of areas.
His interests include the scaling of applications to large processor
counts and system sizes, and the efficient and scalable evaluation of long
ranged interactions, particularly the Coulomb interaction, in such
applications.
Tel: +44 (0) 1865 511245
Email: Ian.Bush@nag.co.uk
Address:
Numerical Algorithms Group Ltd., Wilkinson House, Jordan Hill Road, Oxford, OX2
8DR, United Kingdom
Dr.
Ilian T. Todorov has an MSc in Nuclear Physics (1996, University of Sofia) and
a PhD in Computational Chemistry (2002, University of Bristol). He is a molecular dynamics developer
(DL_POLY) and practitioner in the HPC engineering area. His scientific interests include materials
modelling of non-stoichiometric solid solutions, radiation damage in solid
matter and ion-channel dynamics.
Tel: (+44) 0 1925 60 3681
Email: ilian.todorov@stfc.ac.uk
Dr.
William Smith has a BSc in Chemistry (1972, University of Wales, Cardiff) and a
PhD in Molecular Orbital Theory (1976, University of Wales). He is a Visiting Professor at Sheffield and
Bradford Universities. Molecular
dynamics software developer for parallel computers (DL_POLY etc). His scientific interests include materials
modelling, solutions, polymers, biosystems, semiclassical dynamics, phase
transitions in solids, fast ion conductors and accelerated dynamics methods.
Tel: (+44) 0 1925 60 3257
Email: bill.smith@stfc.ac.uk
Address: CSE Department, STFC Daresbury Laboratory,
Daresbury Science & Innovation Campus, Keckwick Lane, Daresbury, Warrington
WA4 4AD, Cheshire, United Kingdom