Parallelisation
of the adaptive kinetic Monte Carlo code, DL_AKMC
David
S.D. Gunn and John A. Purton
Scientific Computing Department
Science and Technology Facilities Council
Daresbury Laboratory, Sci-Tech Daresbury
Warrington, WA4 4AD
30th
September 2013
Abstract
The
energy and force calculations in the adaptive kinetic Monte Carlo program,
DL_AKMC, have been parallelised using MPI. A task-farm framework has been
incorporated into the code to increase the efficiency of saddle-point evaluation. A confidence limit in the number of saddle
points found has been included, and a variety of new potentials have also been
added to increase the efficiency of the kinetic Monte Carlo algorithm, and to
make the code relevant for a wide selection of materials and systems.
Table of Contents
1.
Background.......................................................................................................................... 3
2.
The dCSE Project and Objectives...................................................................................... 4
2.1
Parallel execution and improved efficiency of the saddle point searches........................ 5
2.2
Improving the efficiency of the energy/force calculations............................................... 6
2.3
Support for a variety of potential types............................................................................ 9
2.4
Defect detection algorithm............................................................................................. 10
2.5
Final reporting................................................................................................................. 11
3.
Conclusions......................................................................................................................... 11
4.
Acknowledgements............................................................................................................. 12
5.
References........................................................................................................................... 12
1. Background
DL_AKMC is a universally
applicable adaptive kinetic Monte Carlo (aKMC) program and is of use to a wide
variety of disciplines. Kinetic Monte
Carlo (KMC)1 techniques are typically used to access long simulation
timescales (up to the order of seconds). KMC constructs a list of possible system
transition events (such as oxygen diffusion) and a move is made on every
iteration of the KMC loop. The list of events can either be created prior to
the simulation or calculated on the fly and we employ the latter approach since
not all the events and defect configurations can be determined prior to the KMC
simulation. Unfortunately, this makes the simulation more demanding since it is
necessary to reliably determine possible events and the activation energies of
these events and the time required to calculate these is significant. We have
developed the methodology to examine diffusion of ionic materials over a few
milliseconds in systems containing a few thousand ions (the overall simulation
time is naturally system dependent). DL_AKMC was originally serial, and
calculated the saddle points and activation energies sequentially. Under this dCSE grant, DL_AKMC has now been
parallelised so that it can make use of the multicore architecture on HECToR
and other HPC systems during the force/energy calculations.
The parallelisation has
focussed on two main areas:
- Saddle point searches
can now be performed on different nodes.
- Force/Energy
calculations are parallelised with a combination of OpenMP and MPI.
DL_AKMC is applicable
to many different areas of science. For
example - the modelling of oxide ion diffusion and consequent ionic
conductivity in fuel cell materials; investigating surface diffusion on ice
mantles; determining healing timescales of materials damaged by radiation. The program is flexible and new methods for
the calculation of activation energies can easily be incorporated and/or
alternate methods for the calculation of the energies of the ions.
Our group, in
collaboration with groups at Bristol and Sheffield universities, is currently employing
atomistic computer simulations to identify suitable ceramic compounds for the
storage of radioactive materials. Radioactive waste disposal is one of the
greatest environmental challenges of the new century2. The present
total volume of radioactive waste in the UK is approximately 3.4 million m3
of which 1,100 m3 is high level waste (HLW)3. Moreover, the legacy of the Cold War requires
the 'neutralisation' of approximately 1500 tonnes of Pu (half life of 239Pu
is 24,100 years). The Committee on Radioactive Waste Management (CoRWM)
recommendation of geological disposal of HLW4 has been accepted by
the Government and has made the problem of safe disposal pressing.
Safe geological
disposal requires that the radionuclides be bound in an inert matrix and buried
deep below the Earth’s surfaces. This matrix must be capable of retaining its
structural integrity during prolonged heavy particle bombardment at
temperatures of 300-600 K. Also any host
material must be resistant to radiation damage and leaching over timescales
comparable to the half-life of the radionuclides.
Radiation damage events
are modelled using a two step process. The first step employs molecular
dynamics (MD) to “shoot” an energetic particle through the host material
creating a cascade (ions are knocked away from their ideal lattice sites). The
second step is to analyse the ability, if any, of the material to heal
(diffusion of ions from defects back to their lattice sites). The latter
process may take place over several seconds and is not amenable to study using
MD (which is limited to timescales typically of the order ~1 ns). Instead, we
use DL_AKMC.
2. The dCSE Project and Objectives
The code developed is
an adaptive Kinetic Monte Carlo (aKMC) code. The aKMC method uses saddle-point
searches and harmonic transition state theory to model rare-event,
state-to-state dynamics in systems of interest. The dynamical events can be
complex, involve many atoms and are not constrained to a grid – relaxing many
of the conditions of regular KMC – making the code more versatile and useful in
situations where the state transitions are not known a priori.
An aKMC simulation
starts form an initially minimised configuration. From this state and all
distinct states visited in the dynamics, saddle point searches are used to find
the processes available to the system. For these searches we use the 'dimer'
method5 as implemented in DL_FIND6, which can generally
find saddle points within a few hundred energy evaluations. We are presently
using the dimer method as it is robust and relatively efficient at calculating
saddle point energies. However, the program has a modular construction and alternative
methods can be included at a later stage without major reconstruction of the
program. The dimer searches were initially consecutive, which was inefficient
as there are no dependencies between saddle-point searches. As a result of this
dCSE grant, the saddle point searches can now be distributed over multiple
nodes, so that saddle point searches can be performed in parallel and
independently of each other. Once all the saddle-point searches are completed
the rates of the corresponding transitions are found using harmonic transition
state theory, and the KMC algorithm advances the system to the next state,
where the process is repeated. A
comparison between the original (serial) and parallel versions of DL_AKMC is
shown by the flowcharts in Figure 1.
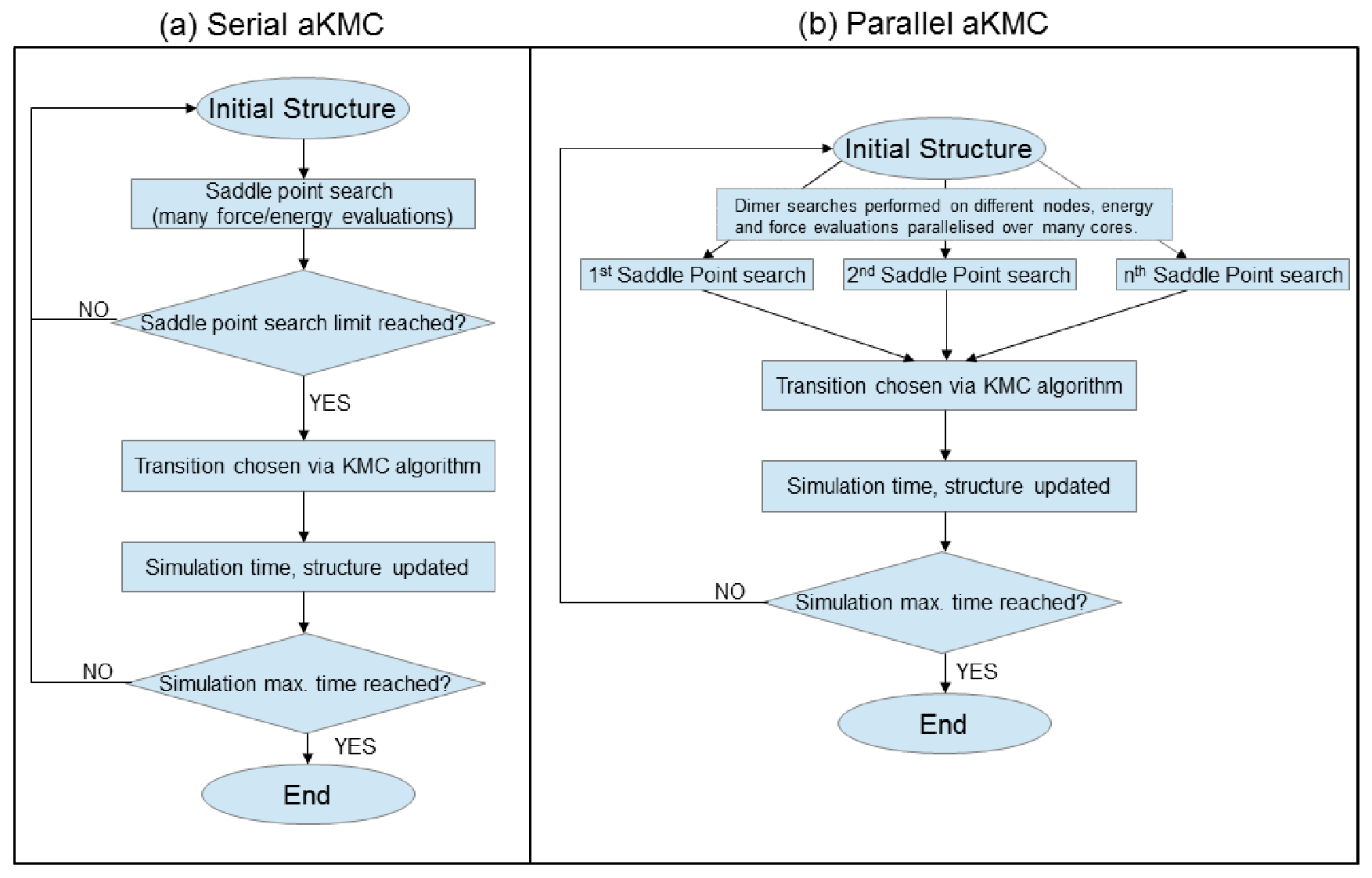
Figure
1:
Workflow of the (a) serial aKMC program and (b) parallel aKMC program, with
parallel distribution of saddle point searches and parallelisation of
force/energy evaluations for each saddle point.
2.1 Parallel execution and improved efficiency of the saddle-point searches
Within a simulation
cell the calculation of the activation energies of different transitions is
achieved either by selecting atoms at random from the bulk mixture, selecting
specific atoms from the bulk mixture, or activating the entire simulation cell
prior to a dimer search. Originally DL_AKMC performed a set number of dimer
searches for each state as defined by the user. This was typically 20-50 dimer
searches for a system of approximately 1000 atoms. This process has now been
improved with work packages (WP) 1.1 and 1.2.
WP 1.1:
Distribution of saddle point searches over different nodes
In the serial program
(Figure 1 (a)), saddle-point searches were performed over a loop with the next
search not starting until the previous one is complete. As each search is
independent, the efficiency can be greatly improved by distributing each saddle
point search over a different node. We have implemented this “task farming”
strategy by splitting the communicator7. The amount of data
transferred between the nodes is small (the positions at the beginning and end
of a saddle point search) and the overhead for communication is relatively
small when compared to the time taken calculating the energies and forces.
Milestone 1.1:
Demonstrate speed up gained by parallelisation of saddle point searches by
comparing serial performance against dimer-parallelised performance for a small
(~2k atoms) radiation-damaged system. 20
dimer searches will be performed for each system state, and the parallel code
will be run on 1, 2, 4, 16 and 32 cores of HECToR. The total simulation time reached after 12
hours will be compared.
Outcome 1.1: Saddle points can now be distributed
over different nodes – as defined by the user in the control file. The metrics for this milestone are somewhat
spurious, as you naturally achieve a 100% (or very near) speedup with each
additional core, as each core is performing its own independent saddle point
search.
WP 1.2:
Confidence limit for the number of dimer searches
To minimise the number
of dimer searches we have incorporated a confidence level for each state that
can describe the probability that an important saddle point has been
missed. This confidence level provides a
dynamic criterion to decide when sufficient saddle-point searches have been
made and therefore speed up the code by eliminating unnecessary saddle-point
searches. Suitable algorithms have been proposed by Xu and Henkelman8.
Milestone 1.2:
Demonstrate the speed up gained by the implementation of the confidence limit
for the saddle point searches, using the system described in Milestone 1.1, and
comparing the total simulation time reached for the fixed 20-dimer searches per
system state (12 hours, 5 nodes of HECToR), with that attained with the
confidence limit imposed under the same conditions.
Outcome 1.2:
The confidence limit has been implemented and a user can specify in the control
file the level of confidence required before the system will progress to the
next state. The implementation of the
confidence limit does not inherently speed up the code, instead it gives a
certain confidence that the simulation is performing at a specified level of
accuracy. If the user specified, for
example, only 5 dimer searches to be performed for each saddle point the
simulation would proceed quickly (compared to one with a high confidence
limit), but the kinetics would be questionable.
2.2 Improving the efficiency of the energy/force calculations
Each saddle point
search requires multiple computations of the system energy and of the forces on
the atoms/ions (approximately 100 computations per saddle-point search) and is
the most time consuming part of the calculation. As part of the dCSE grant we have taken advantage
of the multicore architecture of HECToR and have modified DL_AKMC so that the
energies/forces are calculated on at least one node using a combination of
OpenMP and MPI.
WP 2.1:
Incorporate OpenMP/MPI in the energy/force calculations
The energy and force
calculations primarily run over several nested ‘DO’-loops, which evaluate
functions based upon the distance between an atom and every other atom in the
system. This process is repeated over
every atom in the system to determine the total system energy and individual
forces on each atom. This section of the
code has been made more efficient by parallelisation via replicated data using
OpenMP and MPI, and has made the code suitable for efficiently investigating
systems larger than ~1000 atoms. Each dimer search can be distributed to a
node(s) on HECToR (or other HPC system), and the energy/force evaluations can
be distributed over each core by a combination of OpenMP and MPI. An additional
improvement not originally planned as part of this grant has also been included
– that of a neighbour list. This
increases the efficiency of the force/energy calculations by establishing a
list of neighbours for each atom so that evaluations over the entire cell can
be minimised.
Milestone 2.1:
Demonstrate the speed up gained with the OpenMP/MPI implementation in the
force/energy calculations by comparing the total simulation time reached for
the system in Milestone 1.2 (confidence limit, parallel dimer searches, 12
hours, 5 nodes HECToR), with the simulation time reached under the same
conditions and with the OpenMP/MPI implementation included.
Outcome 2.1:
OpenMP and MPI have been incorporated in the force/energy calculations,
currently MPI-only is default. Due to the random nature of the code total
simulation time comparison is not the best means of comparison. Instead, we have compared the speed up between
a serial calculation and parallel calculations in evaluating 200 energy and
force evaluations of a system (Gd2Ti2O7) that
has ~2000 ions. The parallel
calculations use 2, 4, 8, 16 and 32 cores for each force/energy evaluation. Results
are shown in Figure 2.
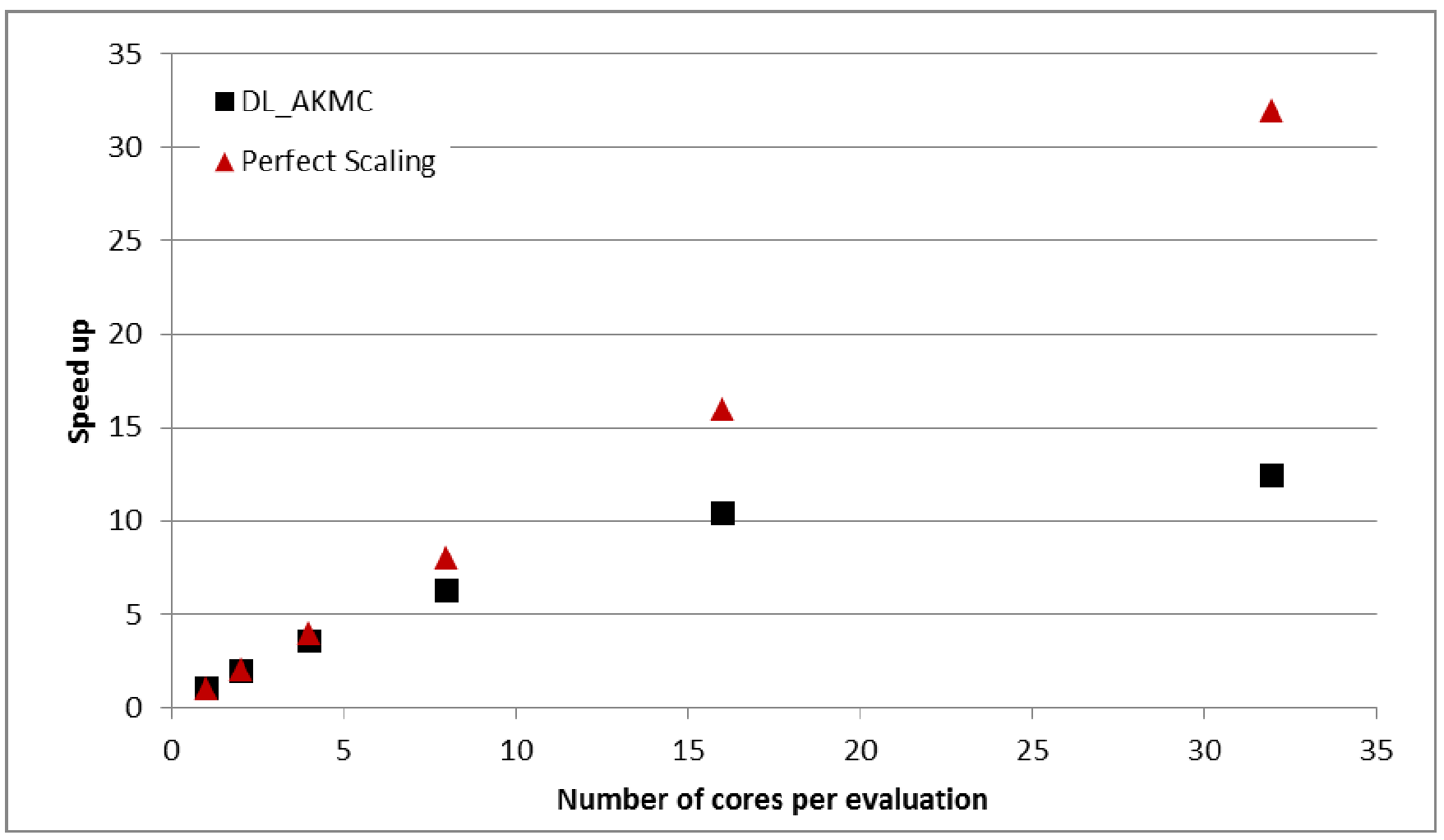
Figure
2:
Speed up between serial and parallel calculations for 200 force/energy
evaluations in a ~2000 ion Gd2Ti2O7 system.
From Figure 2 we can
see that the parallelisation of the force/energy evaluations in DL_AKMC scales well
up to 8 cores, at which point communication overheads slow down the
simulation. This is useful to know, as
the setup can be optimised such that dimer searches can be task-farmed to
blocks of 8 cores to maximise the number and efficiency of dimer searches for a
given number of available cores. We have identified many areas in which this
parallelisation and scalability can be improved, however we lacked time to
implement these under this grant.
Milestone 2.2:
Demonstrate scalability of the code by comparing simulations of different
radiation-damaged system sizes (~2k, 20k, 200k atoms) over different numbers of
cores on HECToR (1, 128, 1024, 2048, 4096, 8192). Scalability will be
determined by comparing the total simulation time reached after 12 hours in
each case.
Outcome 2.2:
DL_AKMC is designed to be used in such a way that maximises both the number of
dimer searches that can be performed in parallel, and the number of cores used
per energy/force evaluation. Measuring
the scalability over a small number of cores is therefore more relevant than
investigating the scalability of performing single dimer searches over a large
number of cores. Milestone 2.2 was
modified to reflect this by comparing the time taken to evaluate 200 energy and
force evaluations of Gd2Ti2O7 systems that
have ~2k, ~20k and ~200k ions with 2, 4, 8, 16 and 32 cores. The results are shown in Figure 3.
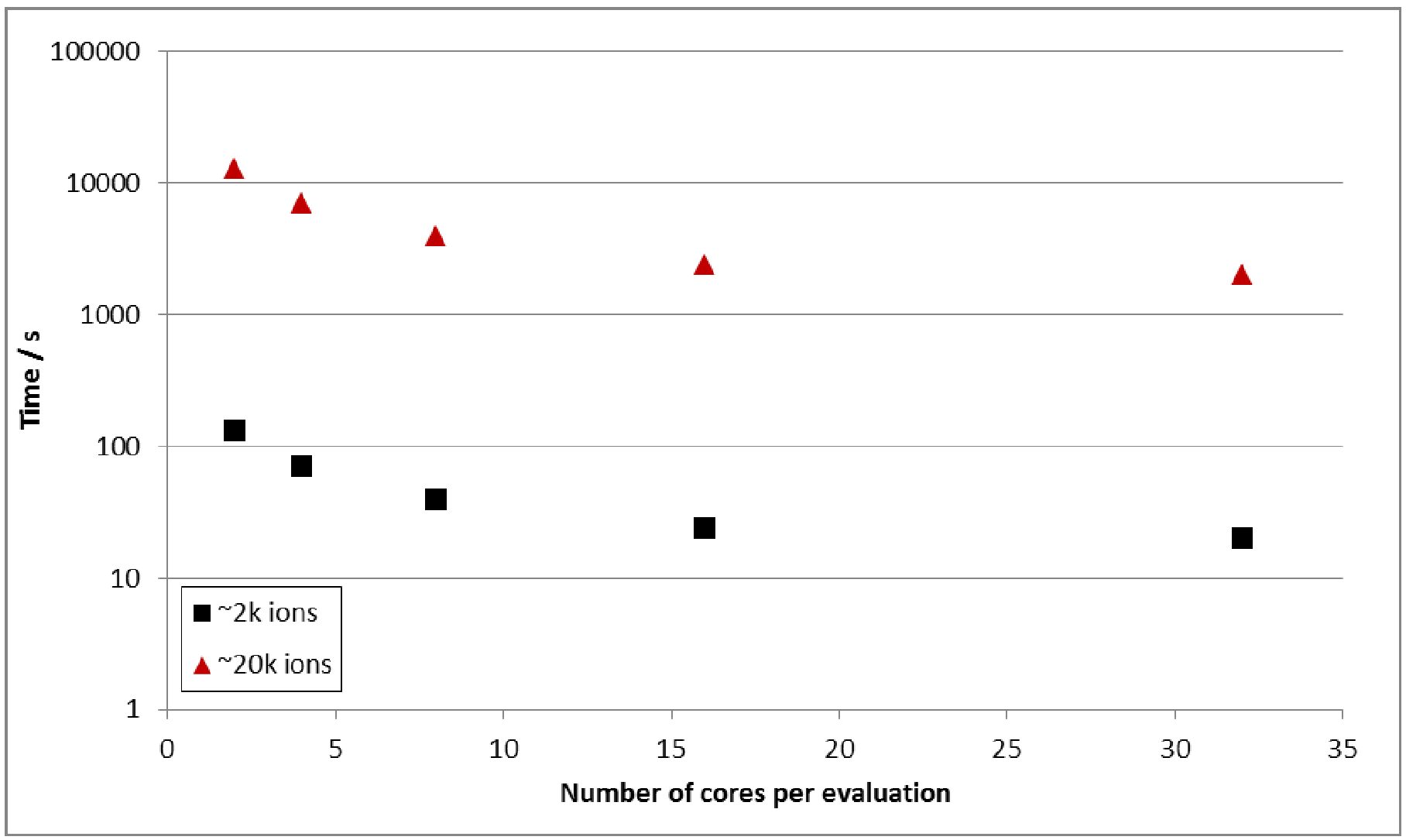
Figure 3:
Time taken for 200 force/energy
evaluations in a ~2k and ~20k ion Gd2Ti2O7
systems.
Simulations of the
~200k ion system did not complete 200 energy/force evaluations within the 12
hour time limit on the queue and so no data for this system is shown in Figure
3. Scalability appears to be
approximately of O(n2).
2.3 Support for a variety of potential types
The code originally
only supported one type of short ranged potential (Buckingham) to describe the
2-body interaction between ions in the system. We have now added a more diverse
suite of potentials – including Embedded Atom Method and Tersoff potentials, substantially
increasing the usefulness of the code to a wider range of users.
WP 3.1:
Tersoff potential
The Tersoff potential
is a special example of a density dependent potential, which has been designed
to reproduce the properties of covalent bonding in systems containing carbon,
silicon, germanium etc. and alloys of these elements. A special feature of the potential is that it
allows bond breaking and associated changes in bond hybridisation. This
potential is of great use for the semi-conductor modelling community.
Milestone 3.1:
Demonstrate the working Tersoff potential by comparing the system energy and
forces calculated for a 512 atom carbon (diamond) system with the DL_AKMC and
with DL_POLY.
Outcome 3.1:
This has been done, and the energy and forces match between DL_AKMC and DL_POLY
Classic.
WP 3.2:
Embedded atom method (EAM)
The EAM potential is a
density dependent potential derived from density functional theory and
describes the bonding of a metal atom ultimately in terms of the local
electronic density. This type of
potential is especially useful in describing the properties of metals and metal
alloys and therefore of use to a wide selection of researchers.
Milestone 3.2:
Demonstrate the working EAM potential by comparing the system energy and forces
calculated for a 256 atom aluminium system with the aKMC code and with DL_POLY.
Outcome 3.2:
This has been done, and the energy and forces match between DL_AKMC and DL_POLY
Classic.
2.4 Defect detection algorithm
Within a simulation
cell the calculation of the activation energies of different transitions is
achieved either by selecting atoms at random from the bulk mixture, selecting
specific atoms from the bulk mixture, or activating the entire simulation cell
prior to a dimer search. These methods have obvious impact on the number of
dimer searches required and hence the time taken for simulation as a whole.
Therefore, intelligent selection of ions at this stage in the program could
dramatically reduce the number of saddle point searches and computational wall
time.
The lowest energy
atomic transitions are typically those located around defective areas in a
material. To speed up an aKMC
calculation the saddle-point searches can be localised around such areas to
find a high proportion of low-energy transitions, which would be the most
likely to occur in any given KMC step.
In a large system (many thousands of atoms), with many randomised
defective areas (as you would have from a sample damaged by radiation) such
areas are hard to pinpoint. A suitable defect detection algorithm is required
that can automate the process of identifying defective areas of the material
and then proceed with a localised saddle-point search.
WP 4.1:
Include Steinhardt order parameters
We had proposed to use
the method developed by Allan and co-workers to identify defective environments
in pyrochlores, as this would greatly speed up the simulation of healing in
such materials and would also give us a means to identify low energy
transitions. The method is based on the
calculation of local bond order parameters based on spherical harmonics (also
known as Steinhardt order parameters9), and has been adapted from
similar approaches used to analyse the structure of crystalline clusters, soft
particle systems and supercooled liquids.
Milestone 4.1:
Demonstrate speed up gained from the implementation of the Steinhardt order
parameters to target local defective sites, by comparing the total simulation
time reached for the system in Milestone 2.1 (OpenMP/MPI, confidence limit,
parallel dimer searches, 12 hours, 5 nodes HECToR), with the simulation time
reached under the same conditions and with the Steinhardt order parameter
implementation included.
Outcome 4.1: We have been unable to implement the
Steinhardt order parameters as we intended.
The algorithm is complex and the method could not be refined enough to
be robust and general purpose in the time available. We are keen to pursue this improvement and
intend to include it in a later release of the code. Once it was identified that this work package
was unachievable in the time available, effort was focussed on other areas of
improvement such as including a neighbour list and increasing the general
efficiency and usability of the code.
2.5 Final reporting
WP 5.2:
Produce user manual for aKMC program
Milestone 5.2:
User manual written and available online.
Outcome 5.2:
The user manual will be available on the CCPForge website (http://ccpforge.cse.rl.ac.uk/gf/project/kmc/
).
3. Conclusions
The substantial
improvement of DL_AKMC via parallel execution of processes to determine
transition states and the incorporation of OpenMP and MPI in the energy/force
calculations has extended both the length and timescales that are able to be
studied by this technique and this has had an immediate benefit in our research
on radiation damage in materials. The damage is created using molecular
dynamics (MD) and large system sizes are a prerequisite, as this helps
eliminate cascades interacting with the periodic image and sound waves from
travelling through the periodic boundary conditions. MD cannot be used to study
the subsequent healing of the material and the availability of DL_AKMC for use
with larger systems enables us to take input from MD simulations rather than
concentrating on model systems. Therefore, the parallelisation of DL_AKMC
allows the healing of these large systems to be modelled on a realistic
timescale.
DL_AKMC will be of wide
use to many members of the materials science modelling community, especially
with the additional inclusion of a more diverse suite of potentials. The
versatility and universal applicability of the code will help researchers in
e.g. modelling diffusion through different electrolyte materials for fuel cell
applications, looking at the recombination of adatoms on a surface for
catalysis, thin film growth or even simulating diffusion in interstellar ices. In the near future, DL_AKMC will be a vital
part of EPSRC grant EP/K016288/1, modelling materials of interest to the energy
sector. DL_AKMC is available under a GNU Lesser General Public Licence (LGPL),
enabling interested members of the community to download and modify the code
for non-commercial use.
4. Acknowledgements
This project was funded
under the HECToR Distributed Computational Science and Engineering (CSE)
Service operated by NAG Ltd. HECToR – A Research Councils UK High End Computing
Service – is the UK’s national supercomputing service, managed by EPSRC on
behalf of the participating Research Councils. Its mission is to support
capability science and engineering in UK academia. The HECToR supercomputers are managed by UoE
HPCx Ltd. and the CSE Support Service is provided by NAG Ltd. http://www.hector.ac.uk
.
5. References
[1] M.R. Sorenson and
A.F. Voter, J. Chem. Phys. 112 (2000) 9599
[2] Royal Society
Policy Document 24/07 Strategy options for the UK’s separated plutonium: http://royalsociety.org/displaypagedoc.asp?id=27169
[3] Nuclear
Decommissioning Authority report, NDA/RWMD/003 (2008)
[4] Committee on
Radioactive Waste Management (CoRWM) report 700: Recommendations to Government.
[5] G. Henkelman and H.
Jónsson, J. Chem. Phys. 111 (1999) 7010
[6] J. Kästner, J.M.
Carr, T.W. Keal, W. Thiel, A.Wander and P. Sherwood, J. Phys. Chem. A 113
(2009) 11856
[7] W. Gropp, E. Lusk
and A. Skjellum, Using MPI: Portable Parellel Programming with Message Passing
Interface, MIT Press (1999) page 61
[8] L. Xu and G.
Henkelman, J. Chem. Phys. 129 (2008) 114104
[9] P. Steinhardt, D.R.
Nelson and M. Ronchetti, Phys. Rev. B 28 (1983) 784
[10] A.F. Voter, Phys.
Rev. B34 (1986) 6819